Clear Sky Science · en
Novel universal domain-centric method for protein classification
Why sorting proteins matters for health
Inside every cell, thousands of tiny protein machines keep life running smoothly. Among the most important are protein kinases, enzymes that switch other proteins on or off and are prime targets for many modern drugs, especially cancer therapies. But scientists still struggle to neatly sort all kinases into families that reflect how they work. This article presents a new way to classify kinases—and, in principle, many other proteins—by focusing on the shared core region that does the job, and on the basic physical and chemical features of its building blocks. That promises sharper maps of protein families and, ultimately, better clues for designing medicines.
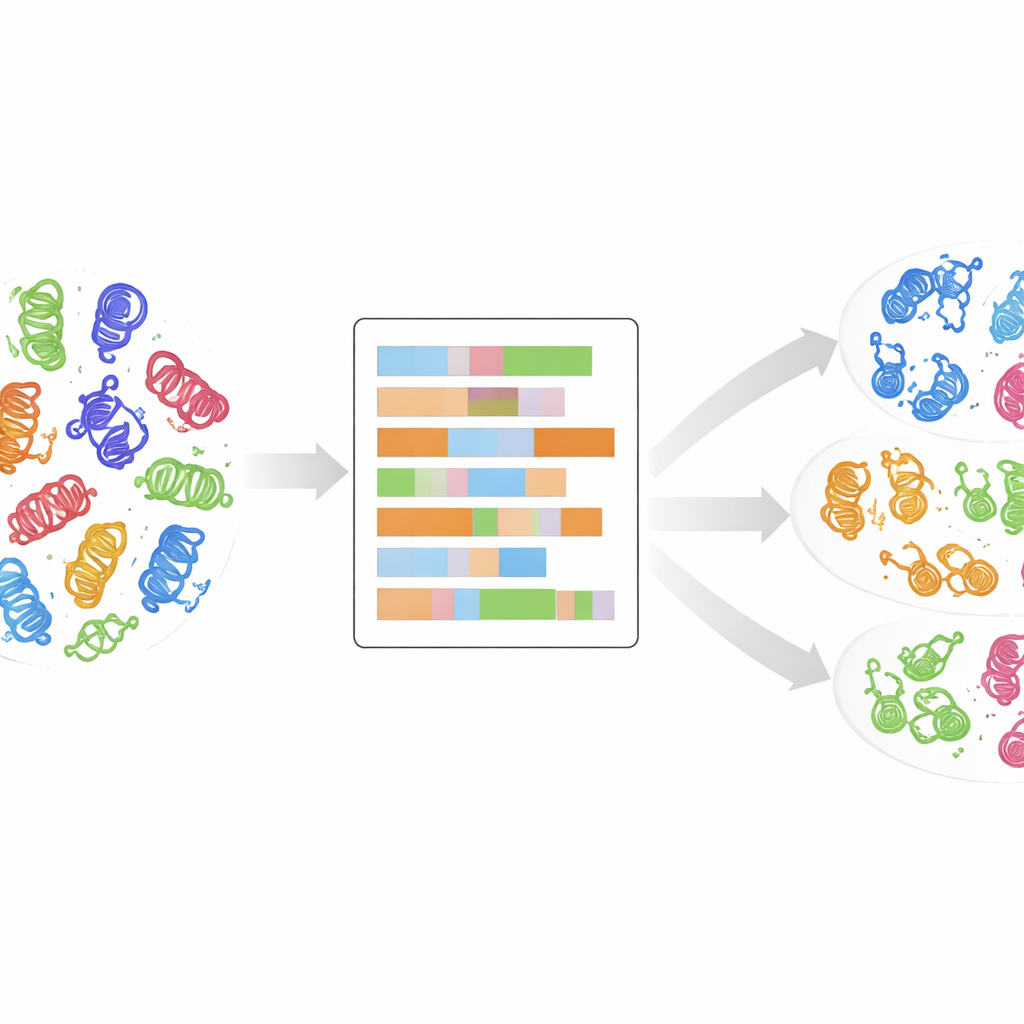
From family trees to a more detailed fingerprint
Traditionally, kinases have been grouped into classes by comparing their gene or protein sequences and building evolutionary “family trees.” This approach has been very successful, revealing about 500 human kinases that fall into several main groups such as AGC, CAMK, CMGC, STE, TK, TKL, and others. Yet many newly discovered kinases do not fit cleanly into these groups: their sequences look different, even though they may behave similarly in cells. Standard comparisons also pay little attention to the underlying properties of each amino acid—the size, charge, or water-loving character that determine how a protein folds and functions. The authors argue that to truly understand kinase families, we need to look beyond alphabet-like sequence matching and examine these physico-chemical fingerprints.
Zooming in on the active core of kinases
To do this, the researchers used a high-quality alignment of 497 human kinase domains, the compact cores that actually carry out the chemical reaction of adding phosphate groups. For every position in this shared domain map, they replaced the amino acid letter with up to 30 numerical descriptors that capture properties such as charge, hydrophobicity, polarity, and size, plus an extra marker for gaps. The result is a detailed numerical portrait of each kinase domain, where similar behavior in three-dimensional space should translate into similar patterns in these numbers. They then reduced the complexity of these portraits using principal component analysis, a standard technique that condenses many measurements into a few main directions capturing the largest differences.
Letting the data cluster itself
Without telling the computer which kinase belonged to which known class, the team applied an unsupervised clustering method called k-means to the reduced numerical data. They explored many possible numbers of clusters and used statistical scores to identify the most meaningful groupings, then combined several such solutions into a final set of 24 clusters, each with a confidence score reflecting how stable it was across runs. Remarkably, about 90% of kinases ended up in clusters that matched their original class labels, suggesting that the physico-chemical domain portrait naturally recovers—and sometimes sharpens—existing classifications. Some clusters contained a mix of a major class and previously “OTHER” kinases, hinting that those outliers may in fact belong to an established family.
Discovering key structural hot spots
Beyond grouping, the method reveals which parts of the kinase domain actually drive these differences. By combining the principal components with the residue properties and then shuffling data in randomization tests, the authors pinpointed specific positions whose property patterns strongly distinguish one class from the others. One standout example is a site in the activation loop region of CMGC kinases that almost always carries a positively charged residue, unlike most other classes. Structural models show that in a representative CMGC kinase, this residue helps stabilize nearby phosphorylated sites that are crucial for turning the enzyme on. Interestingly, an “unclassified” kinase called CDC7 shares a similar environment at this site, supporting the prediction that it behaves like a CMGC kinase even if its evolutionary history is different.
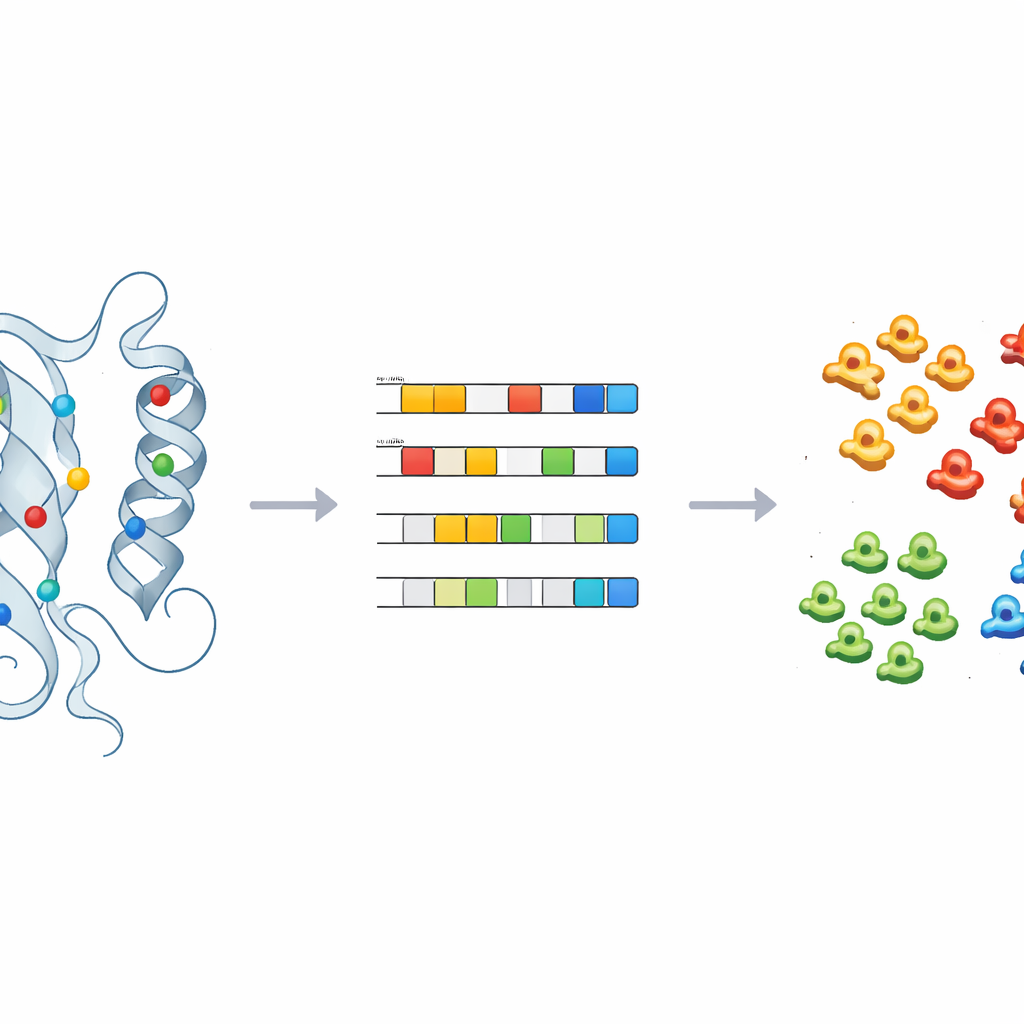
Teaching machines to label the unknown
To turn these insights into practical predictions, the team trained supervised machine learning models—including logistic regression, random forests, and a probabilistic classifier—on the physico-chemical fingerprints of kinases with known labels. After careful tuning and cross-validation, these models could accurately assign kinases to main classes using only a handful of principal components. Applied to 66 kinases that had been left in the catch-all “OTHER” group, the models consistently re-assigned several, such as CDC7 and members of the ULK family, to specific kinase classes. Structural checks of these reassignments, especially around the key positions identified earlier, supported the machine predictions and illustrated how the method can guide reclassification and experimental follow-up.
A general recipe for mapping protein families
In everyday terms, this work shows that proteins can be sorted not just by spelling out their sequences, but by distilling how their core parts behave physically and chemically. For kinases, this domain-centric, property-based view recovers known families, helps re-label misfits, and highlights structural “hot spots” that matter for activity and regulation. Because the recipe relies only on a shared domain alignment and general amino acid descriptors, the authors also demonstrate that it can be applied to other protein groups, such as small GTPases, and could extend to immunoglobulins, G protein–coupled receptors, and beyond. As such maps become more refined, they could steer the design of more selective drugs, aid in interpreting disease-causing mutations, and offer a clearer, function-focused atlas of the protein universe.
Citation: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Keywords: protein kinases, protein classification, machine learning, protein domains, structure-based biology