Clear Sky Science · ru
Новый универсальный метод классификации белков, ориентированный на домены
Почему важна сортировка белков для здоровья
В каждой клетке тысячи крошечных белковых машин поддерживают жизнь. Среди наиболее значимых — протеинкиназы, ферменты, которые включают или выключают другие белки и служат главными целями для многих современных лекарств, особенно против рака. Тем не менее учёным по-прежнему трудно аккуратно распределить все киназы по семействам, которые отражали бы их функционирование. В этой статье представлен новый способ классификации киназ — и, в принципе, многих других белков — с акцентом на общую ключевую область, выполняющую работу, и на базовые физико-химические свойства её строительных блоков. Это обещает более чёткие карты семейств белков и, в конечном счёте, лучшие подсказки для разработки препаратов.
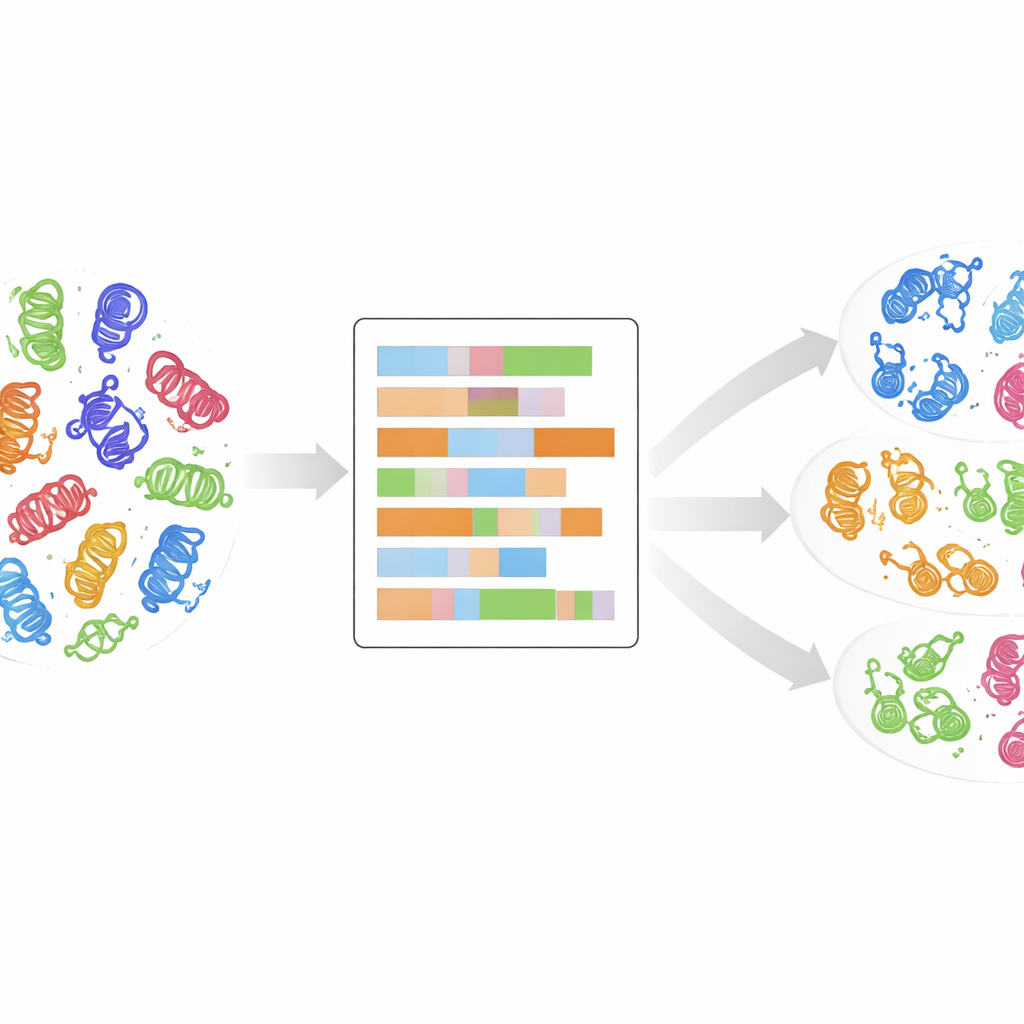
От филогенетических деревьев к более детальному отпечатку
Традиционно киназы группируют, сравнивая их генетические или белковые последовательности и строя эволюционные «семейные деревья». Этот подход дал большие результаты, выявив около 500 человеческих киназ, которые делятся на несколько основных групп, таких как AGC, CAMK, CMGC, STE, TK, TKL и другие. Однако многие недавно обнаруженные киназы не вписываются чётко в эти группы: их последовательности выглядят иначе, хотя в клетке они могут вести себя похоже. Стандартные сравнения также мало учитывают базовые свойства каждой аминокислоты — размер, заряд или гидрофильность, определяющие, как белок сворачивается и функционирует. Авторы утверждают, что для глубокого понимания семейств киназ нужно смотреть дальше буквального сопоставления последовательностей и исследовать эти физико‑химические отпечатки.
Прицел на активное ядро киназ
Для этого исследователи использовали высококачественное выравнивание 497 доменов человеческих киназ — компактных ядер, которые непосредственно осуществляют химическую реакцию присоединения фосфатных групп. Для каждой позиции в этой общей карте домена они заменили буквенное обозначение аминокислот на до 30 числовых дескрипторов, отражающих такие свойства, как заряд, гидрофобность, полярность и размер, а также добавили маркер для пропусков. В результате получился детализированный числовой портрет каждого домена киназы, где сходное поведение в трёхмерном пространстве должно переводиться в похожие закономерности в этих числах. Затем они уменьшили размерность этих портретов с помощью анализа главных компонент — стандартной техники, конденсирующей большое число измерений в несколько направлений, которые улавливают наибольшие различия.
Позволив данным самим образовывать кластеры
Не сообщая компьютеру, к какой известной классовой метке принадлежит каждая киназа, команда применила метод неконтролируемой кластеризации k‑means к уменьшенным числовым данным. Они исследовали множество вариантов числа кластеров и использовали статистические коэффициенты для выбора наиболее значимых группировок, затем объединили несколько таких решений в итоговый набор из 24 кластеров, у каждого из которых есть оценка доверия, отражающая стабильность при повторных запусках. Поразительно, примерно 90% киназ оказались в кластерах, соответствующих их исходным классам, что говорит о том, что физико‑химический доменный портрет естественно восстанавливает — а иногда и уточняет — существующие классификации. Некоторые кластеры сочетали основную группу и ранее отнесённые к «OTHER» киназы, что намекает, что эти выбросы на самом деле могут принадлежать уже установленному семейству.
Открытие ключевых структурных «горячих точек»
Помимо группировки, метод выявляет, какие участки домена киназы действительно определяют эти различия. Комбинируя главные компоненты с дескрипторами остатков и выполняя тесты с рандомизацией, авторы выделили конкретные позиции, шаблоны свойств которых сильно отличают один класс от остальных. Яркий пример — сайт в активационной петле CMGC‑киназ, который почти всегда содержит положительно заряженную аминокислоту, в отличие от большинства других классов. Структурные модели показывают, что в представительной CMGC‑киназе этот остаток помогает стабилизировать соседние фосфорилированные участки, критичные для активации фермента. Интересно, что «неклассифицированная» киназа CDC7 имеет похожую среду в этом месте, что поддерживает предположение о её поведении как CMGC‑киназы, даже если эволюционная история отличается.
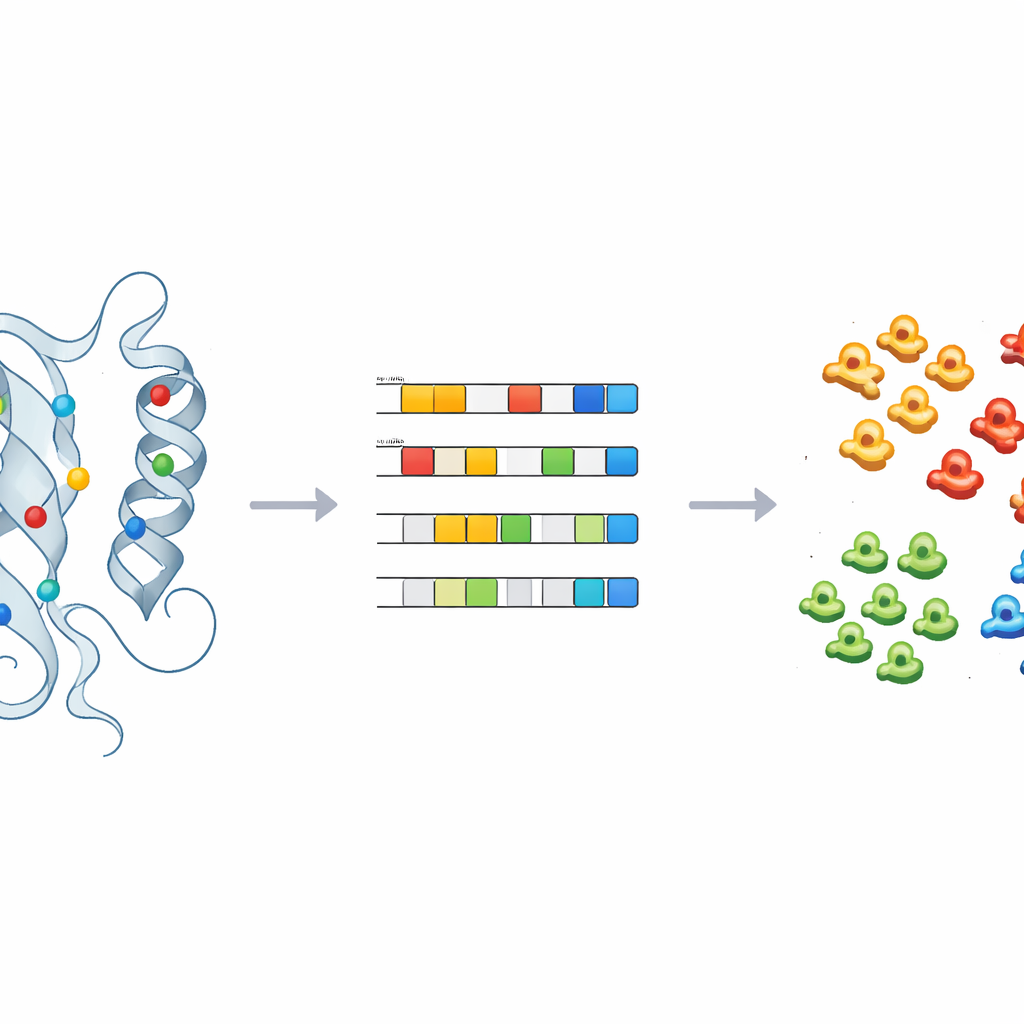
Обучение машин помечать неизвестное
Чтобы превратить эти наблюдения в практические предсказания, команда обучила контролируемые модели машинного обучения — включая логистическую регрессию, случайные леса и вероятностный классификатор — на физико‑химических отпечатках киназ с известными метками. После тщательной настройки и перекрёстной проверки эти модели могли точно относить киназы к основным классам, используя лишь несколько главных компонент. Применённые к 66 киназам, оставленным в обобщённой группе «OTHER», модели последовательно переназначили несколько белков, таких как CDC7 и члены семейства ULK, в конкретные классы киназ. Структурные проверки этих переназначений, особенно вблизи ранее идентифицированных ключевых позиций, подтвердили предсказания машин и показали, как метод может направлять реклассификацию и последующие экспериментальные проверки.
Универсальный рецепт для картирования семейств белков
Проще говоря, эта работа демонстрирует, что белки можно сортировать не только по «буквам» их последовательностей, но и по тому, как их ключевые части ведут себя физически и химически. Для киназ такой ориентированный на домен и свойство подход восстанавливает известные семейства, помогает переназначать аномалии и выделяет структурные «горячие точки», важные для активности и регуляции. Поскольку метод опирается лишь на общее выравнивание домена и универсальные дескрипторы аминокислот, авторы показывают, что его можно применять и к другим группам белков, таким как малые GTP‑азы, а также расширять на иммуноглобулины, G‑белок‑связанные рецепторы и далее. По мере того как такие карты станут более точными, они могут направлять разработку более селективных лекарств, помогать в интерпретации вызывающих болезни мутаций и давать более ясную, ориентированную на функцию карту белкового мира.
Цитирование: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Ключевые слова: протеинкиназы, классификация белков, машинное обучение, домены белков, структурная биология