Clear Sky Science · pt
Novo método universal centrado em domínios para classificação de proteínas
Por que classificar proteínas importa para a saúde
Dentro de cada célula, milhares de pequenas máquinas proteicas mantêm a vida funcionando sem sobressaltos. Entre as mais importantes estão as quinasas de proteína, enzimas que ligam ou desligam outras proteínas e são alvos principais de muitos medicamentos modernos, especialmente terapias contra o câncer. Ainda assim, os cientistas têm dificuldade em agrupar todas as quinasas em famílias que reflitam seu modo de ação. Este artigo apresenta uma nova forma de classificar quinasas — e, em princípio, muitas outras proteínas — ao focar na região core compartilhada que realiza o trabalho, e nas características físicas e químicas básicas de seus blocos construtores. Isso promete mapas mais nítidos das famílias proteicas e, em última instância, pistas melhores para o desenvolvimento de medicamentos.
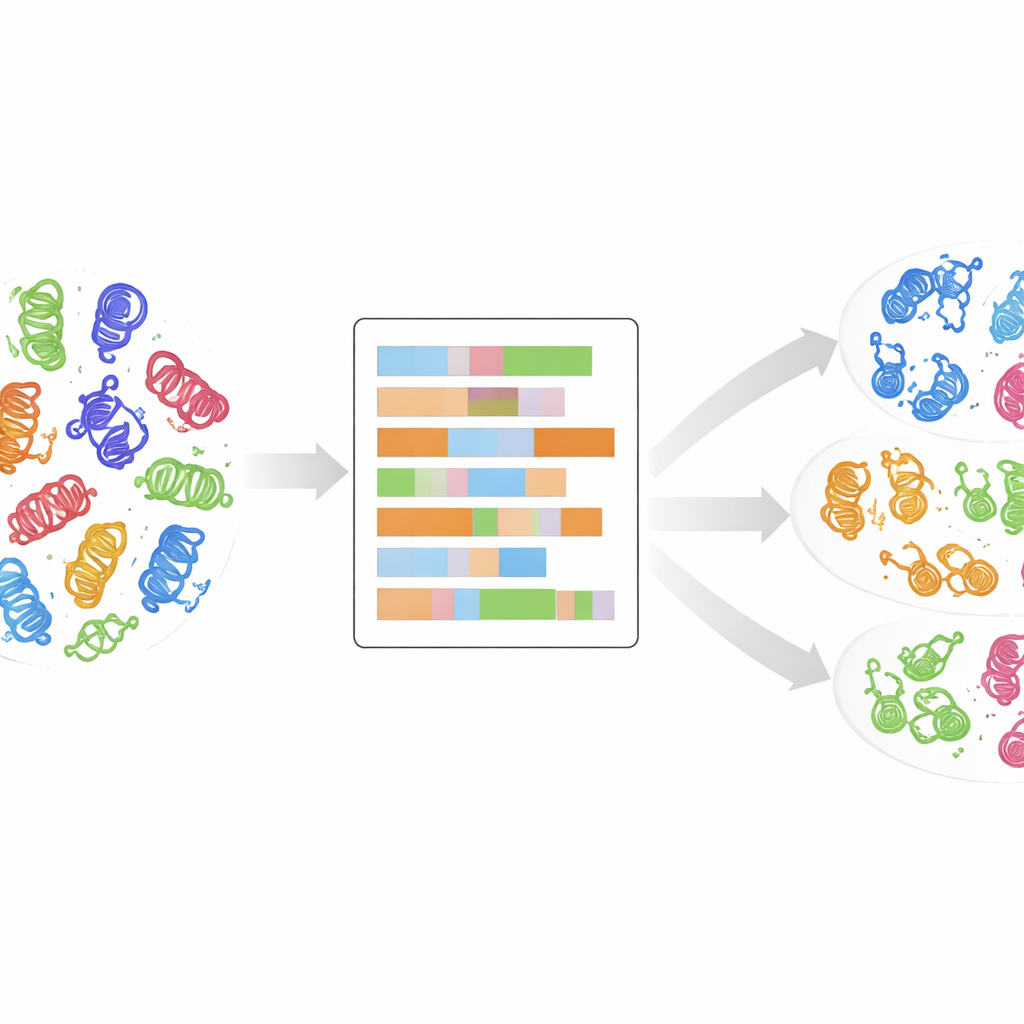
Das árvores genealógicas a uma impressão digital mais detalhada
Tradicionalmente, as quinasas têm sido agrupadas em classes comparando suas sequências gênicas ou proteicas e construindo “árvores” evolutivas. Essa abordagem teve grande sucesso, revelando cerca de 500 quinasas humanas que se encaixam em vários grupos principais como AGC, CAMK, CMGC, STE, TK, TKL e outros. Ainda assim, muitas quinasas recém-descobertas não se encaixam bem nesses grupos: suas sequências parecem diferentes, mesmo que possam se comportar de modo semelhante nas células. Comparações padrão também dão pouca atenção às propriedades subjacentes de cada aminoácido — o tamanho, carga ou caráter hidrofílico que determinam como uma proteína se dobra e funciona. Os autores argumentam que, para entender de fato as famílias de quinasas, precisamos olhar além do pareamento das sequências como se fosse um alfabeto e examinar essas impressões digitais físico-químicas.
Aproximando-se do núcleo ativo das quinasas
Para isso, os pesquisadores usaram um alinhamento de alta qualidade de 497 domínios de quinases humanas, os núcleos compactos que realmente realizam a reação química de adicionar grupos fosfato. Para cada posição nesse mapa de domínio compartilhado, eles substituiram a letra do aminoácido por até 30 descritores numéricos que capturam propriedades como carga, hidrofobicidade, polaridade e tamanho, além de um marcador extra para lacunas. O resultado é um retrato numérico detalhado de cada domínio de quinase, onde comportamentos similares no espaço tridimensional devem se traduzir em padrões semelhantes nesses números. Em seguida, reduziram a complexidade desses retratos usando análise de componentes principais, uma técnica padrão que condensa muitas medições em algumas direções principais que capturam as maiores diferenças.
Deixando os dados se agruparem sozinhos
Sem informar ao computador a qual classe conhecida cada quinase pertencia, a equipe aplicou um método de agrupamento não supervisionado chamado k-means aos dados numéricos reduzidos. Eles exploraram muitos números possíveis de clusters e usaram métricas estatísticas para identificar os agrupamentos mais significativos, depois combinaram várias dessas soluções em um conjunto final de 24 clusters, cada um com uma pontuação de confiança que reflete quão estável foi entre execuções. Notavelmente, cerca de 90% das quinasas terminaram em clusters que correspondiam às suas etiquetas de classe originais, sugerindo que o retrato do domínio baseado em propriedades físico-químicas recupera — e às vezes afina — as classificações existentes. Alguns clusters continham uma mistura de uma classe principal e quinasas antes rotuladas como “OUTRAS”, insinuando que esses outliers podem, de fato, pertencer a uma família estabelecida.
Descobrindo pontos estruturais-chave
Além do agrupamento, o método revela quais partes do domínio de quinase realmente impulsionam essas diferenças. Combinando os componentes principais com as propriedades dos resíduos e depois embaralhando os dados em testes de randomização, os autores identificaram posições específicas cujos padrões de propriedades distinguem fortemente uma classe das demais. Um exemplo marcante é um sítio na região do loop de ativação das quinasas CMGC que quase sempre carrega um resíduo positivamente carregado, diferentemente da maioria das outras classes. Modelos estruturais mostram que, em uma quinase representativa da família CMGC, esse resíduo ajuda a estabilizar sítios fosforilados próximos que são cruciais para ativar a enzima. Curiosamente, uma quinase “não classificada” chamada CDC7 compartilha um ambiente semelhante nesse sítio, apoiando a previsão de que ela se comporta como uma quinase CMGC mesmo que sua história evolutiva seja diferente.
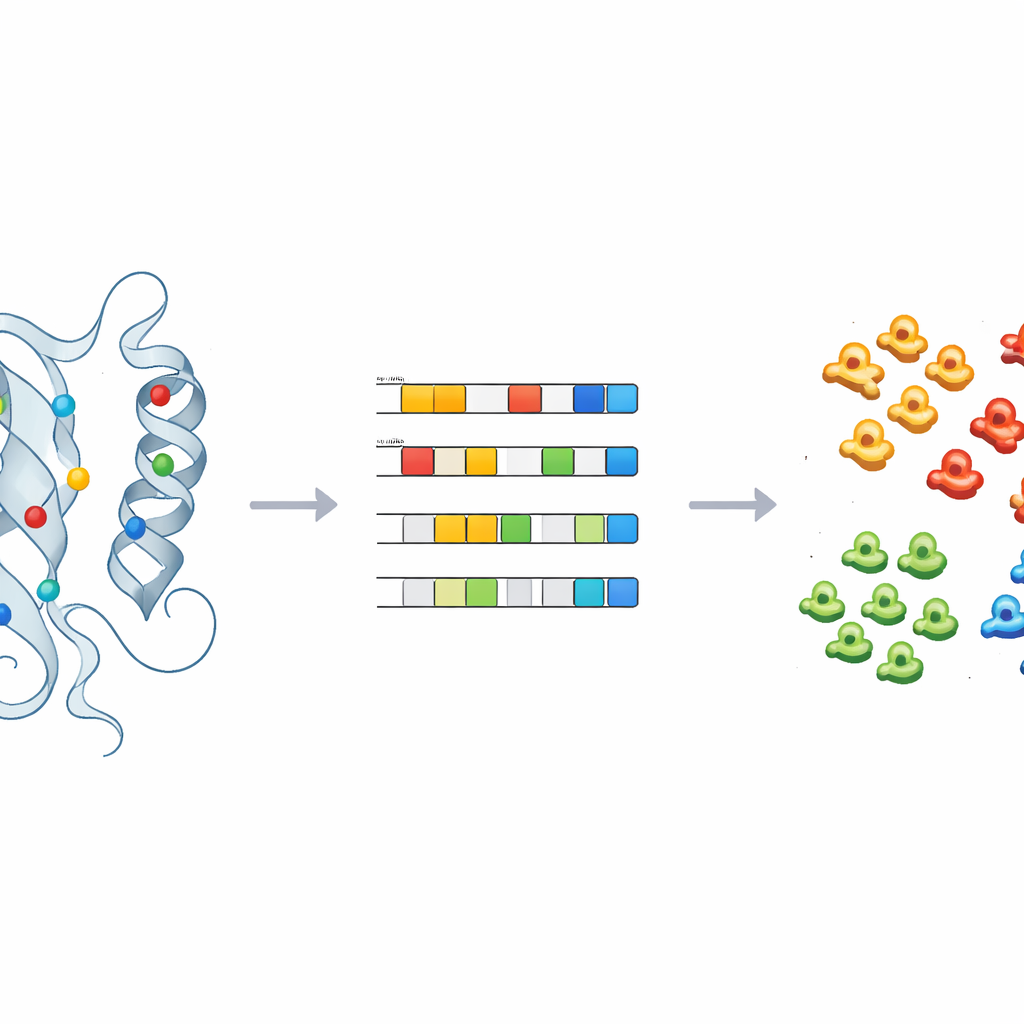
Ensinando máquinas a rotular o desconhecido
Para transformar esses insights em previsões práticas, a equipe treinou modelos supervisionados de aprendizado de máquina — incluindo regressão logística, florestas aleatórias e um classificador probabilístico — nos retratos físico-químicos das quinasas com rótulos conhecidos. Após ajuste cuidadoso e validação cruzada, esses modelos puderam atribuir com precisão quinasas às classes principais usando apenas um punhado de componentes principais. Aplicados a 66 quinasas que haviam sido deixadas no grupo genérico “OUTRAS”, os modelos reatribuíram de forma consistente várias delas, como CDC7 e membros da família ULK, para classes específicas de quinasas. Verificações estruturais dessas reatribuições, especialmente ao redor das posições-chave identificadas anteriormente, apoiaram as previsões das máquinas e ilustraram como o método pode orientar reclassificações e investigações experimentais subsequentes.
Uma receita geral para mapear famílias de proteínas
Em termos práticos, este trabalho mostra que proteínas podem ser ordenadas não apenas pelo soletrar de suas sequências, mas por destilar como suas partes centrais se comportam fisicamente e quimicamente. Para as quinasas, essa visão centrada no domínio e baseada em propriedades recupera famílias conhecidas, ajuda a reclassificar os desalinhados e destaca “pontos quentes” estruturais que importam para atividade e regulação. Como a receita depende apenas de um alinhamento de domínio compartilhado e de descritores gerais de aminoácidos, os autores também demonstram que ela pode ser aplicada a outros grupos de proteínas, como pequenas GTPases, e pode se estender a imunoglobulinas, receptores acoplados a proteínas G e além. À medida que tais mapas se refinam, eles podem orientar o desenho de fármacos mais seletivos, auxiliar na interpretação de mutações causadoras de doenças e oferecer um atlas mais claro e focado na função do universo proteico.
Citação: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Palavras-chave: quinasas de proteína, classificação de proteínas, aprendizado de máquina, domínios de proteínas, biologia baseada em estrutura