Clear Sky Science · nl
Nieuwe universele domeingerichte methode voor eiwitclassificatie
Waarom het sorteren van eiwitten belangrijk is voor de gezondheid
In elke cel zorgen duizenden kleine eiwitmachientjes ervoor dat het leven soepel verloopt. Tot de belangrijkste behoren proteïnekinasen, enzymen die andere eiwitten aan- of uitzetten en die veel doelwitten zijn voor moderne medicijnen, vooral bij kankertherapieën. Toch hebben wetenschappers nog moeite om alle kinasen netjes in families in te delen die weerspiegelen hoe ze werken. Dit artikel presenteert een nieuwe manier om kinasen — en in principe veel andere eiwitten — te classificeren door te focussen op het gedeelde kerngebied dat het werk doet en op de fundamentele fysisch-chemische eigenschappen van de bouwstenen. Dat belooft scherpere kaarten van eiwitfamilies en uiteindelijk betere aanwijzingen voor geneesmiddelenontwerp.
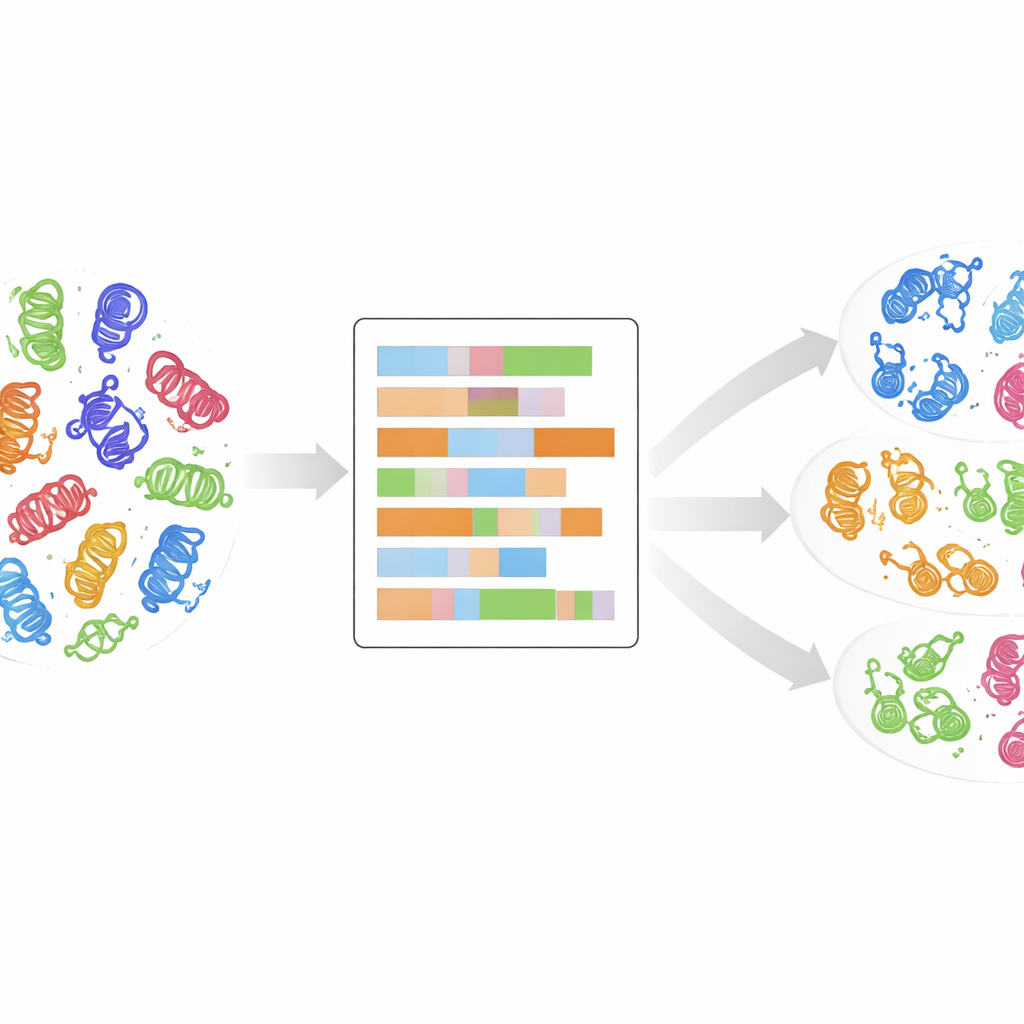
Van stambomen naar een gedetailleerder vingerafdruk
Traditioneel worden kinasen in groepen ingedeeld door hun gen- of eiwitsequenties te vergelijken en evolutionaire “stambomen” te construeren. Deze aanpak was zeer succesvol en bracht ongeveer 500 menselijke kinasen aan het licht die vallen in enkele hoofdgroepen zoals AGC, CAMK, CMGC, STE, TK, TKL en andere. Toch passen veel recent ontdekte kinasen niet netjes in deze groepen: hun sequenties zien er anders uit, ook al kunnen ze zich in cellen vergelijkbaar gedragen. Standaardvergelijkingen houden ook weinig rekening met de onderliggende eigenschappen van elk aminozuur — de grootte, lading of hydrofiele aard die bepalen hoe een eiwit vouwt en functioneert. De auteurs stellen dat om kinase-families echt te begrijpen, we verder moeten kijken dan alfabetachtige sequentiewedstrijden en deze fysisch-chemische vingerafdrukken moeten onderzoeken.
Inzoomen op de actieve kern van kinasen
Om dit te doen gebruikten de onderzoekers een hoogwaardige uitlijning van 497 menselijke kinasedomeinen, de compacte kernen die daadwerkelijk de chemische reactie van fosfaattoevoeging uitvoeren. Voor elke positie in deze gedeelde domeinkaart vervingen ze de aminozuurletter door tot 30 numerieke beschrijvers die eigenschappen vastleggen zoals lading, hydrofobiciteit, polariteit en grootte, plus een extra aanduiding voor gaps. Het resultaat is een gedetailleerd numeriek portret van elk kinasedomein, waarbij vergelijkbaar gedrag in driedimensionale ruimte zich zou moeten vertalen in vergelijkbare patronen in deze getallen. Ze reduceerden vervolgens de complexiteit van deze portretten met hoofdcomponentenanalyse, een gangbare techniek die veel metingen samenvat in een paar hoofdrichtingen die de grootste verschillen vangen.
De data zelf laten clusteren
Zonder de computer te vertellen welke kinase tot welke bekende klasse behoorde, paste het team een onbewaakt clusteren toe genaamd k-means op de gereduceerde numerieke data. Ze onderzochten veel mogelijke aantallen clusters en gebruikten statistische scores om de meest betekenisvolle groeperingen te identificeren, waarna ze meerdere van zulke oplossingen combineerden tot een eindset van 24 clusters, elk met een betrouwbaarheidscore die aangeeft hoe stabiel het cluster over runs was. Remarkabel genoeg belandde ongeveer 90% van de kinasen in clusters die overeenkwamen met hun oorspronkelijke klasselabels, wat suggereert dat het fysisch-chemische domeinportret bestaand classificaties natuurlijk herstelt — en soms aanscherpt. Sommige clusters bevatten een mix van een grote klasse en eerder als “OTHER” aangeduide kinasen, wat suggereert dat die uitschieters mogelijk wel degelijk tot een gevestigde familie behoren.
Belangrijke structurele hotspots ontdekken
Buiten het groeperen onthult de methode welke delen van het kinasedomein deze verschillen daadwerkelijk aansturen. Door de hoofdcomponenten te combineren met de residue-eigenschappen en vervolgens data te schudden in randomisatietests, identificeerden de auteurs specifieke posities waarvan de eigenschappatronen sterk één klasse onderscheiden van de anderen. Een opvallend voorbeeld is een plaats in de activatielus van CMGC-kinasen die vrijwel altijd een positief geladen residu draagt, in tegenstelling tot de meeste andere klassen. Structurele modellen tonen dat in een representatieve CMGC-kinase dit residu helpt nabijgelegen gefosforyleerde sites te stabiliseren die cruciaal zijn voor het activeren van het enzym. Interessant genoeg deelt een “niet-geclassificeerde” kinase genaamd CDC7 een vergelijkbare omgeving op deze positie, wat de voorspelling ondersteunt dat het zich gedraagt als een CMGC-kinase, zelfs als zijn evolutionaire geschiedenis anders is.
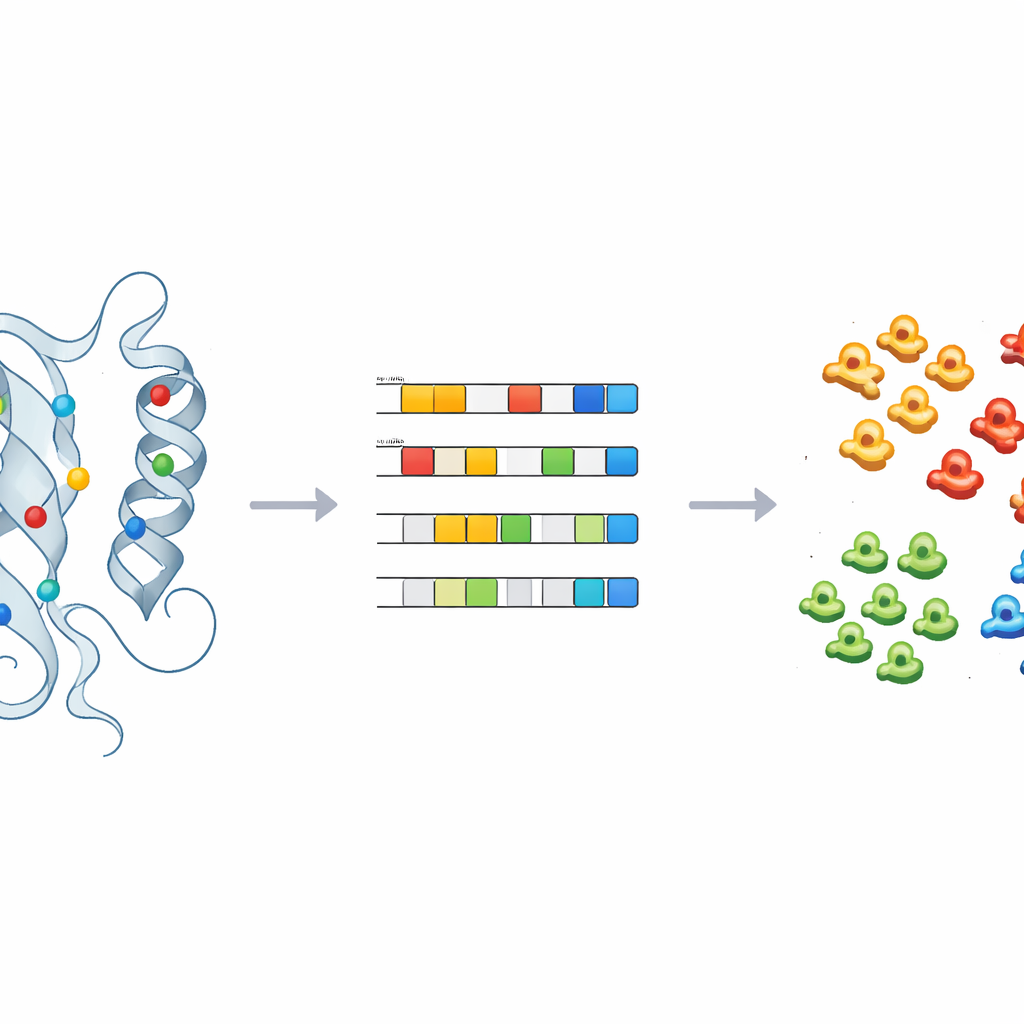
Machines leren de onbekenden te labelen
Om deze inzichten in praktische voorspellingen om te zetten, trainde het team toegezienleerde machine-learningmodellen — waaronder logistieke regressie, random forests en een probabilistische classifier — op de fysisch-chemische vingerafdrukken van kinasen met bekende labels. Na zorgvuldige afstemming en cross-validatie konden deze modellen kinasen nauwkeurig toewijzen aan hoofdklassen met slechts een handvol hoofdcomponenten. Toegepast op 66 kinasen die in de verzamelgroep “OTHER” waren achtergelaten, wezen de modellen consequent meerdere opnieuw toe, zoals CDC7 en leden van de ULK-familie, aan specifieke kinaseklassen. Structurele controles van deze herindelingen, vooral rond de eerder geïdentificeerde sleutelposities, ondersteunden de machinevoorspellingen en illustreerden hoe de methode reclassificatie en experimenteel vervolg kan sturen.
Een algemeen recept voor het in kaart brengen van eiwitfamilies
In gewone bewoordingen laat dit werk zien dat eiwitten niet alleen gesorteerd kunnen worden door hun sequenties te spellen, maar door te destilleren hoe hun kernonderdelen zich fysisch en chemisch gedragen. Voor kinasen herstelt dit domeingerichte, eigenschapsgebaseerde perspectief bekende families, helpt het misfits opnieuw te labelen en belicht het structurele “hotspots” die van belang zijn voor activiteit en regulatie. Omdat het recept alleen steunt op een gedeelde domeinuitlijning en algemene aminozuurbeschrijvers, tonen de auteurs ook aan dat het toepasbaar is op andere eiwitgroepen, zoals kleine GTPases, en zich kan uitstrekken tot immunoglobulinen, G-eiwitgekoppelde receptoren en verder. Naarmate zulke kaarten verfijnder worden, kunnen ze het ontwerp van selectievere medicijnen sturen, helpen bij het interpreteren van ziekteveroorzakende mutaties en een helderder, op functie gericht atlas van het eiwituniversum bieden.
Bronvermelding: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Trefwoorden: proteïnekinasen, eiwitclassificatie, machine learning, eiwitdomeinen, structuurgedreven biologie