Clear Sky Science · sv
Biallelica varianter i den icke-kodande RNA-genen RNU4-2 orsakar ett recessivt neurodevelopmentalt syndrom med karakteristiska förändringar i vitt substans
Varför denna sällsynta genetiska berättelse spelar roll
De flesta av oss tänker på gener som ritningar för proteiner, men vissa gener arbetar tyst i bakgrunden och hjälper celler att redigera andra geners budskap. Denna artikel avslöjar en ny barndomssjukdom i hjärnan som inte orsakas av ett trasigt protein, utan av små förändringar i en icke-kodande RNA-gen kallad RNU4-2. Genom att följa familjer runt om i världen visar forskarna hur ärvda förändringar i denna gen stör hjärnans kopplingar, ger ett igenkännbart mönster på MR-bilder och skiljer sig från en bättre känd, besläktad sjukdom. Arbetet illustrerar hur subtila fel i cellens RNA-redigeringsmaskineri kan omforma hjärnans utveckling — och hur nya genomiska verktyg äntligen blottlägger dessa dolda sjukdomar.
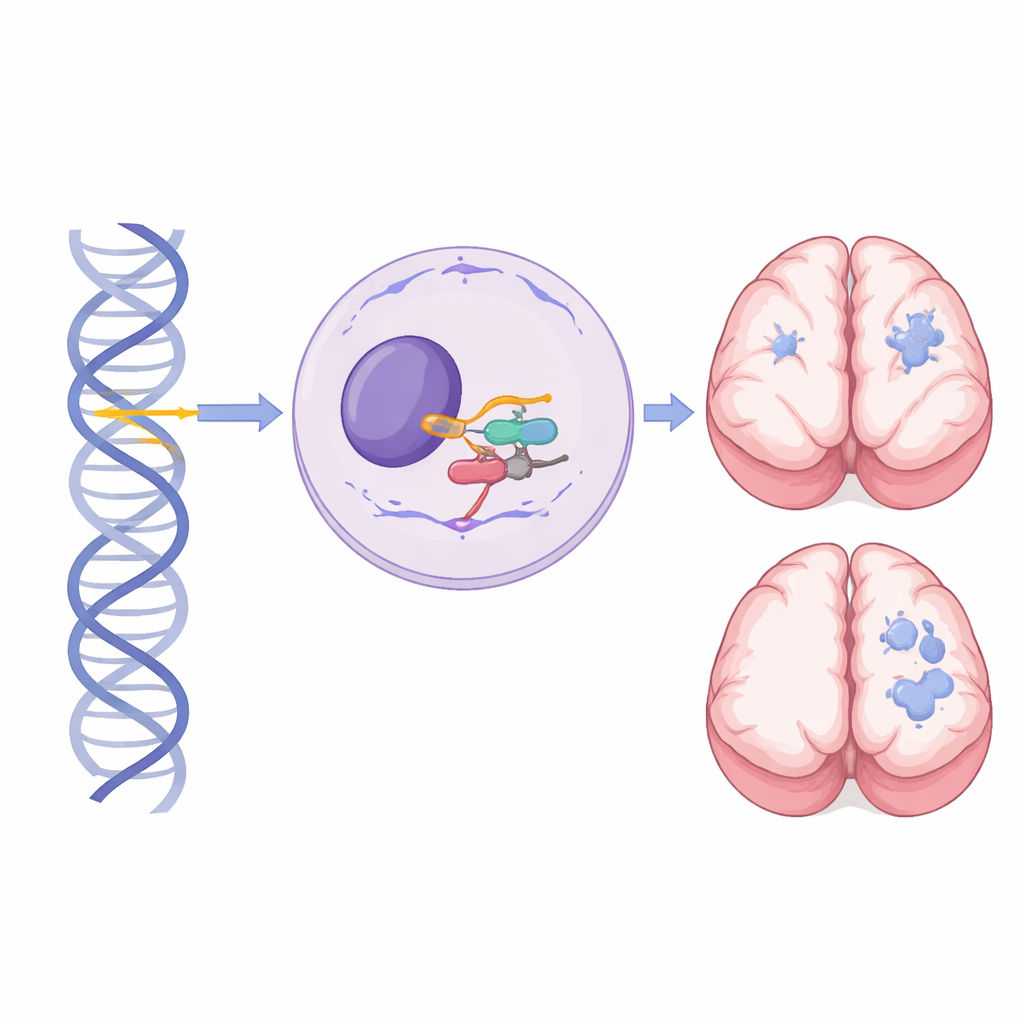
En liten gen med stor betydelse för hjärnans utveckling
Inuti varje cell klipper och klistrar en stor maskin kallad spliceosomen RNA-budskap så att gener kan bli fungerande molekyler. RNU4-2 kodar för U4 small nuclear RNA, en kritisk komponent i den stora spliceosomen, som hanterar nästan alla introner i våra gener. Tidigare har enstaka nya mutationer i ett kort, centralt segment av RNU4-2 visats orsaka en vanlig dominant neurodevelopmental sjukdom känd som ReNU-syndrom: ett barn blir drabbat även om bara en kopia av genen är förändrad. I kontrast fokuserar denna studie på personer som ärvt skadliga varianter i båda kopiorna av RNU4-2 — en från vardera föräldern — och avslöjar ett distinkt, recessivt syndrom.
Att hitta familjer och ett gemensamt kliniskt utseende
Forskargruppen sökte i stora sekvenseringsprojekt för sällsynta sjukdomar och biobanker för att hitta individer med två RNU4-2-varianter och neurodevelopmentala problem. De identifierade slutligen 38 personer från 28 familjer och samlade detaljerad klinisk data för 31 av dem. Nästan alla hade global utvecklingsförsening eller intellektuell funktionsnedsättning, ofta i måttlig till svår grad, med uttalad talförsening; cirka hälften saknade meningsfullt tal. Många barn började gå sent, och mer än en fjärdedel gick aldrig självständigt vid fem års ålder. Låg muskeltonus i spädbarnsåldern, anfall, rörelse- och koordinationproblem samt beteendemässiga utmaningar som aggression eller tvångsmässiga drag var vanliga. Hudförändringar, ögonproblem, kortvuxenhet och genitala avvikelser hos pojkar visade sig hos en betydande minoritet, vilket bildar en multisystembild snarare än en sjukdom som enbart drabbar hjärnan.
Hjärnavbildning avslöjar ett slående vitt substans-signatur
Magnetisk resonansavbildning gav en av de tydligaste ledtrådarna att detta recessiva tillstånd skiljer sig från det dominanta ReNU-syndromet. Bland 27 individer med skanningar eller rapporter hade 24 avvikelser, och en pediatrisk neuroradiolog granskade 13 skanningar i detalj. Varje granskad skanning visade involvering av vitt substans, mest karakteristiskt en utvidgning av de vätskefyllda utrymmen som omger små blodkärl (perivaskulära utrymmen) i det djupa och periventrikulära vita substansen. I de svåraste fallen samlades dessa utrymmen till tätt packade, mikrocystliknande kluster, ofta åtföljda av förtunning av corpus callosum och krympning av lillhjärnan. Detta dramatiska mönster hade inte rapporterats i ReNU-syndromet och sågs sällan hos personer med RNU4-2-varianter i den allmänna populationen, vilket gör det till en kraftfull radiologisk signatur som kan leda till riktad genetisk utredning.
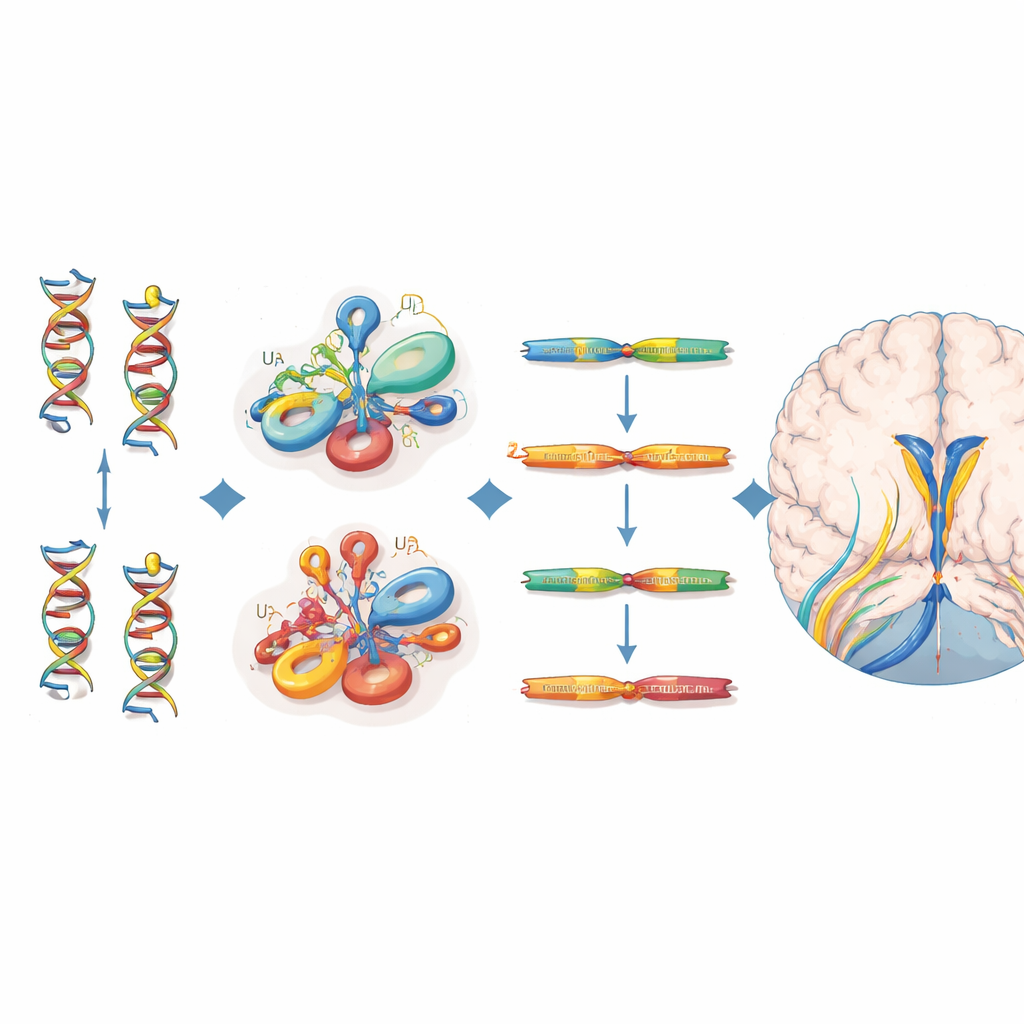
Preciserande av mutationerna och deras molekylära effekt
Alla 38 drabbade bar biallela varianter som klustrades i specifika strukturella regioner av U4-RNA — särskilt Stem II-regionen som binder till U6-RNA, en skarp böj kallad k-turn som rekryterar proteiner, och en sträcka som krävs för bindning av stabiliserande Sm-proteiner. Många av dessa positioner motsvarar ekvivalenta platser i en besläktad RNA, RNU4ATAC, där mutationer redan är kända för att orsaka andra recessiva sjukdomar. För att bedöma hur skadliga varje variant är drog författarna nytta av ett saturation genome editing-experiment som mätt hur hundratals möjliga RNU4-2-förändringar påverkar cellsurvival. Varianter hos patienter med neurodevelopmental sjukdom tenderade att ha lägre funktionspoäng än de som sågs hos friska frivilliga i UK Biobank, vilket stödjer deras roll i sjukdomen, även om inte varje kliniskt viktig variant nådde en strikt gräns i detta cellbaserade test.
Annorlunda sjukdomsmekanism än det dominanta systersyndromet
Genom analys av RNA-sekvenseringsdata visade forskarna att barn med det recessiva RNU4-2-tillståndet har kraftigt minskade nivåer av RNU4-2-RNA, tillsammans med en kraftig ökning av en närbesläktad RNA, RNU4-1. Kvoten mellan RNU4-2 och RNU4-1 var mycket lägre än i tusentals andra patientprover och kunde pålitligt särskilja drabbade individer, vilket tyder på en loss-of-function-mekanism delvis motverkad — men inte helt räddad — av uppreglering av RNU4-1. I kontrast hade personer med dominant ReNU-syndrom faktiskt förhöjda nivåer av RNU4-2 och en karakteristisk förskjutning i hur vissa splice-sites används, vilket pekar på en kvalitativt annorlunda molekylär störning. Noterbart är att trots U4:s centrala roll i den stora spliceosomen upptäckte teamet inte det breda intron-retentionsmönstret som ses i besläktade sjukdomar i den lilla spliceosomen, vilket antyder ett mer subtilt eller vävnadsspecifikt fel i RNA-processningen.
Vad detta betyder för familjer och framtida diagnoser
För icke-specialister är huvudbudskapet att denna studie definierar en ny ärftlig hjärnsjukdom orsakad av att ha två felaktiga kopior av en liten, icke-kodande RNA-gen. Barn med detta tillstånd visar typiskt betydande utvecklingsförsening, frekventa anfall och ett karakteristiskt mönster av förändringar i vitt substans på MR, men de är kliniskt skilda från patienter med det vanligare, dominant verkande ReNU-syndromet. Upptäckten utvidgar det kända spektrumet av sjukdomar kopplade till spliceosomens RNA-komponenter, understryker vikten av att inkludera små RNA-gener i genetiska testpaneler och pekar på enkla biomarkörer — såsom uttrycksrelationen RNU4-2 till RNU4-1 och förekomsten av extrem dilation av perivaskulära utrymmen — som kan hjälpa till att avsluta den diagnostiska odyssén för berörda familjer.
Citering: Rius, R., Blakes, A.J.M., Chen, Y. et al. Biallelic variants in the noncoding RNA gene RNU4-2 cause a recessive neurodevelopmental syndrome with distinct white matter changes. Nat Genet 58, 761–773 (2026). https://doi.org/10.1038/s41588-026-02554-6
Nyckelord: neurodevelopmental störning, spliceosom, icke-kodande RNA, förändringar i vitt substans, RNU4-2