Clear Sky Science · it
Varianti biallelici nel gene dell’RNA non codificante RNU4-2 causano una sindrome neuroevolutiva recessiva con distinti cambiamenti della materia bianca
Perché questa rara storia genetica è importante
La maggior parte di noi pensa ai geni come a progetti per le proteine, ma alcuni geni operano silenziosamente dietro le quinte, aiutando le cellule a modificare i messaggi di altri geni. Questo articolo identifica un nuovo disturbo cerebrale infantile causato non da una proteina difettosa, ma da piccole alterazioni in un gene di RNA non codificante chiamato RNU4-2. Seguendo famiglie in tutto il mondo, i ricercatori mostrano come variazioni ereditarie in questo gene compromettano il cablaggio cerebrale, producano un modello riconoscibile alla risonanza magnetica e si distinguano da una condizione correlata e meglio nota. Il lavoro illustra come difetti sottili nella macchina cellulare che elabora l’RNA possano rimodellare lo sviluppo cerebrale — e come nuovi strumenti genomici stiano finalmente rivelando queste malattie nascoste.
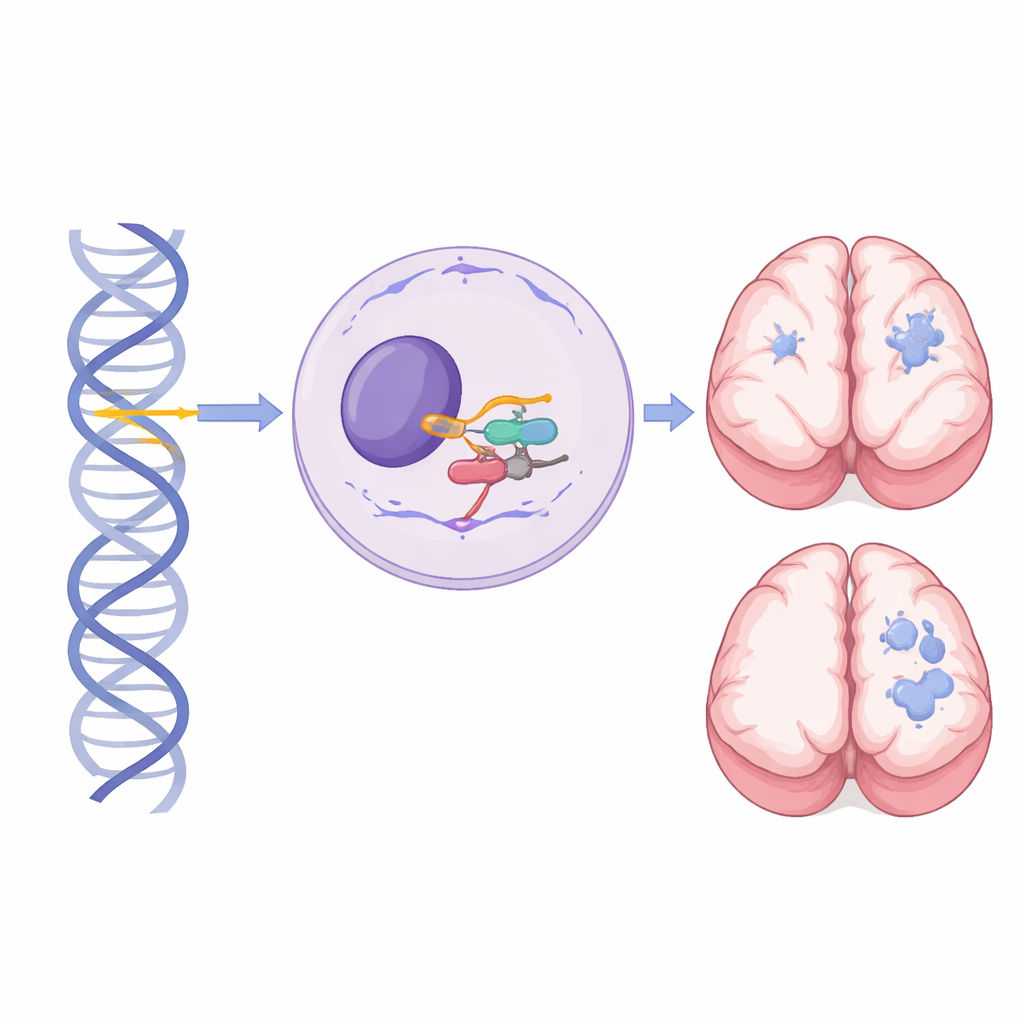
Un piccolo gene con un grande ruolo nello sviluppo cerebrale
All’interno di ogni cellula, una grande macchina chiamata spliceosoma taglia e incolla i messaggi di RNA affinché i geni possano essere trasformati in molecole funzionanti. RNU4-2 codifica l’RNA nucleare piccolo U4, un componente critico del principale spliceosoma, che gestisce quasi tutti gli introni nei nostri geni. In precedenza, mutazioni de novo in un breve tratto centrale di RNU4-2 erano state mostrate responsabili di una condizione neuroevolutiva dominante comune nota come sindrome ReNU: un bambino è affetto anche se è alterata solo una copia del gene. Invece, questo studio si concentra su persone che hanno ereditato varianti dannose in entrambe le copie di RNU4-2 — una da ciascun genitore — rivelando una sindrome recessiva distinta.
Ricerca delle famiglie e quadro clinico condiviso
Il gruppo ha setacciato grandi progetti di sequenziamento per malattie rare e biobanche per trovare individui con due varianti di RNU4-2 e problemi neuroevolutivi. Hanno identificato in totale 38 persone provenienti da 28 famiglie e raccolto dati clinici dettagliati per 31 di loro. Quasi tutti presentavano ritardo globale dello sviluppo o disabilità intellettiva, spesso di grado da moderato a grave, con marcato ritardo del linguaggio; circa la metà non aveva un linguaggio parlato significativo. Molti bambini hanno iniziato a camminare tardi, e oltre un quarto non ha mai camminato in modo indipendente entro i cinque anni. Ipotonia nella prima infanzia, crisi epilettiche, problemi di movimento e coordinazione e difficoltà comportamentali come aggressività o tratti ossessivi erano comuni. Alterazioni della pelle, problemi oculari, bassa statura e anomalie genitali nei maschi comparivano in una minoranza consistente, configurando un quadro multi-sistemico più che una malattia limitata al solo cervello.
Le scansioni cerebrali rivelano una firma marcata sulla materia bianca
La risonanza magnetica ha fornito uno degli indizi più chiari che questa condizione recessiva è distinta dalla sindrome ReNU dominante. Tra 27 individui con scansioni o referti, 24 presentavano anomalie, e un neuroradiologo pediatrico ha esaminato in dettaglio 13 scansioni. Ogni scansione rivista mostrava coinvolgimento della materia bianca, più caratteristicamente un’espansione degli spazi pieni di liquido che circondano i piccoli vasi sanguigni (spazi perivascolari) nella materia bianca profonda e periventricolare. Nei casi più gravi questi spazi si fondevano in ammassi strettamente compatti, simili a microcisti, spesso accompagnati da assottigliamento del corpo calloso e riduzione del cervelletto. Questo pattern drammatico non era stato riportato nella sindrome ReNU ed era raramente osservato in persone con varianti di RNU4-2 dalla popolazione generale, rendendolo una firma radiologica potente che può indirizzare verso test genetici mirati.
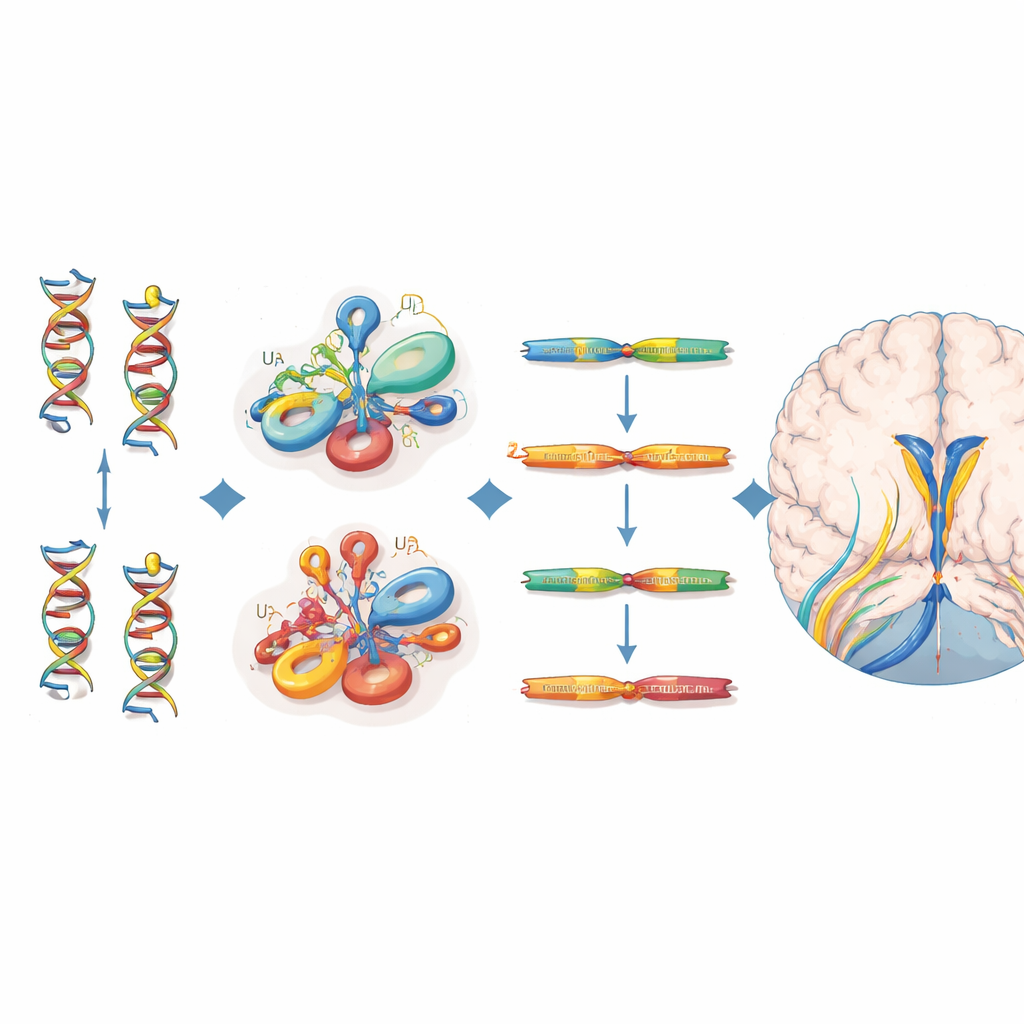
Individuare le mutazioni e il loro impatto molecolare
Tutti i 38 individui colpiti portavano varianti biallelici che si raggruppavano in specifiche regioni strutturali dell’RNA U4 — in particolare la regione Stem II che si lega all’RNA U6, una piega pronunciata chiamata k-turn che recluta proteine, e un tratto necessario per il legame delle proteine Sm stabilizzanti. Molte di queste posizioni corrispondono a siti equivalenti in un RNA correlato, RNU4ATAC, dove mutazioni sono già note per causare altre malattie recessive. Per valutare quanto ogni variante fosse dannosa, gli autori hanno utilizzato un esperimento di saturazione di editing genomico che aveva misurato come centinaia di possibili cambiamenti di RNU4-2 influenzassero la sopravvivenza cellulare. Le varianti nei pazienti con malattia neuroevolutiva tendevano ad avere punteggi di funzione più bassi rispetto a quelle osservate in volontari sani del UK Biobank, sostenendo il loro ruolo nella malattia, anche se non ogni variante clinicamente rilevante soddisfaceva una soglia rigida in questo saggio basato su cellule.
Meccanismo di malattia diverso rispetto alla sindrome sorella dominante
Analizzando dati di sequenziamento dell’RNA, i ricercatori hanno mostrato che i bambini con la condizione recessiva legata a RNU4-2 presentano livelli marcatamente ridotti di RNA RNU4-2, insieme a un netto aumento di un RNA strettamente correlato, RNU4-1. Il rapporto RNU4-2/RNU4-1 era molto più basso rispetto a migliaia di altri campioni di pazienti e poteva distinguere in modo affidabile gli individui affetti, suggerendo un meccanismo di perdita di funzione parzialmente compensato — ma non completamente salvato — dall’aumento di RNU4-1. Al contrario, le persone con la sindrome ReNU dominante avevano in realtà livelli aumentati di RNU4-2 e uno spostamento caratteristico nell’uso di alcuni siti di splicing, indicativo di un disturbo molecolare qualitativamente diverso. È notevole che, nonostante il ruolo centrale di U4 nel principale spliceosoma, il gruppo non abbia rilevato il diffuso schema di ritenzione degli introni osservato in disturbi correlati del minor spliceosoma, implicando un difetto più sottile o specifico per alcuni tessuti nell’elaborazione dell’RNA.
Cosa significa per le famiglie e per le diagnosi future
Per i non specialisti, il messaggio chiave è che questo studio definisce un nuovo disturbo cerebrale ereditario causato dall’avere due copie difettose di un piccolo gene di RNA non codificante. I bambini con questa condizione mostrano tipicamente ritardo dello sviluppo significativo, crisi epilettiche frequenti e un pattern caratteristico di alterazione della materia bianca alla risonanza magnetica, ma sono clinicamente distinti dai pazienti con la sindrome ReNU, che agisce in modo dominante e più comune. La scoperta amplia lo spettro noto di malattie legate ai componenti di RNA dello spliceosoma, sottolinea l’importanza di includere i piccoli geni di RNA nei pannelli diagnostici genetici e indica semplici biomarcatori — come il rapporto di espressione RNU4-2 su RNU4-1 e la presenza di marcata dilatazione degli spazi perivascolari — che possono aiutare a porre fine all’odissea diagnostica per le famiglie colpite.
Citazione: Rius, R., Blakes, A.J.M., Chen, Y. et al. Biallelic variants in the noncoding RNA gene RNU4-2 cause a recessive neurodevelopmental syndrome with distinct white matter changes. Nat Genet 58, 761–773 (2026). https://doi.org/10.1038/s41588-026-02554-6
Parole chiave: disturbo neuroevolutivo, spliceosoma, RNA non codificante, cambiamenti della materia bianca, RNU4-2