Clear Sky Science · pt
Variantes bialélicas no gene de RNA não codificante RNU4-2 causam uma síndrome neurodesenvolvimental recessiva com alterações distintas da substância branca
Por que essa rara história genética importa
A maioria de nós pensa em genes como plantas de construção para proteínas, mas alguns genes trabalham discretamente nos bastidores, ajudando as células a editar as mensagens de outros genes. Este artigo revela um novo distúrbio cerebral infantil causado não por uma proteína defeituosa, mas por pequenas alterações em um gene de RNA não codificante chamado RNU4-2. Ao acompanhar famílias ao redor do mundo, os pesquisadores mostram como alterações herdadas nesse gene perturbam a conexão cerebral, produzem um padrão reconhecível na ressonância magnética e diferem de uma condição relacionada e mais conhecida. O trabalho ilustra como falhas sutis na maquinaria de edição de RNA da célula podem remodelar o desenvolvimento cerebral — e como novas ferramentas genômicas finalmente estão revelando essas doenças ocultas.
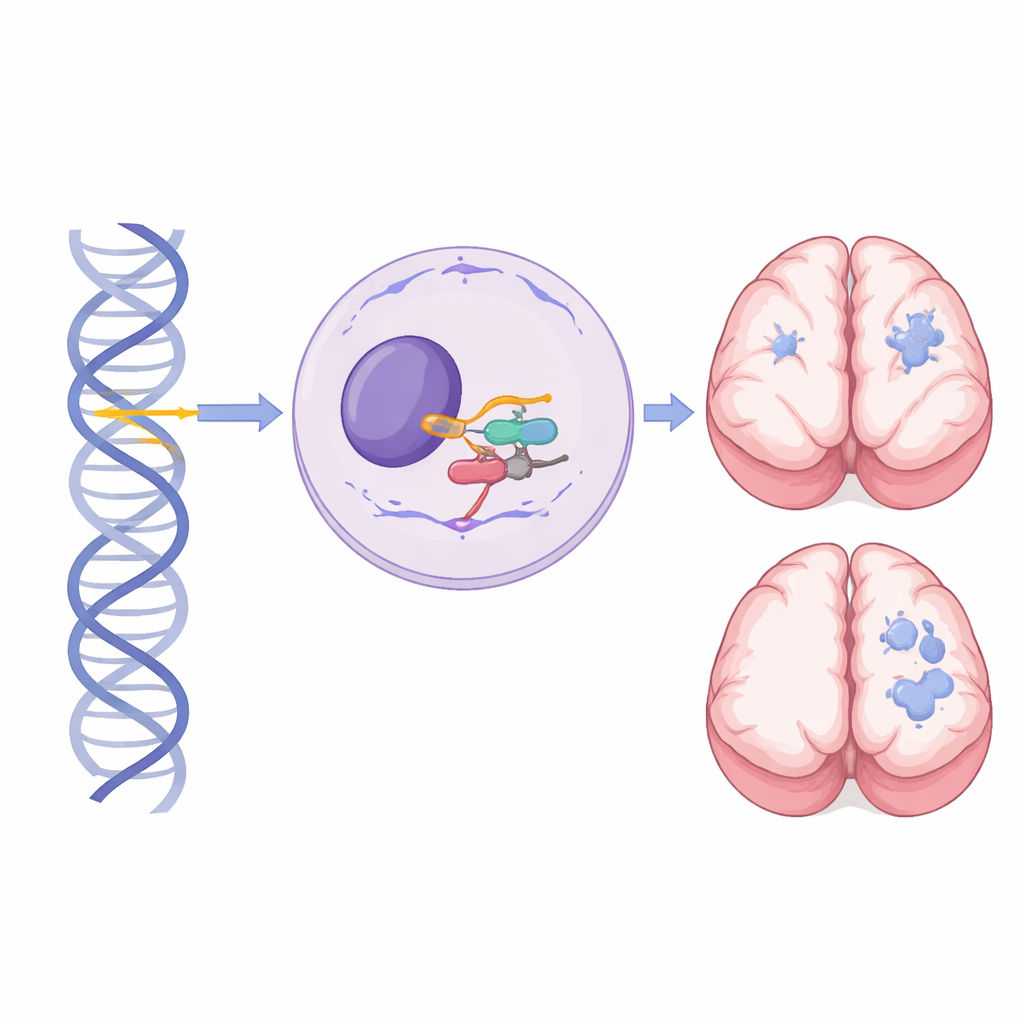
Um gene pequeno com grande papel no desenvolvimento cerebral
Dentro de cada célula, uma grande máquina chamada spliceossomo corta e cola mensagens de RNA para que os genes possam ser transformados em moléculas funcionais. RNU4-2 codifica o RNA nuclear pequeno U4, um componente crítico do spliceossomo maior, que processa quase todos os íntrons dos nossos genes. Anteriormente, mutações de novo em um trecho curto e central de RNU4-2 mostraram causar uma condição neurodesenvolvimental dominante conhecida como síndrome ReNU: a criança é afetada mesmo que apenas uma cópia do gene esteja alterada. Em contraste, este estudo foca em pessoas que herdaram variantes danosas em ambas as cópias de RNU4-2 — uma de cada progenitor — revelando uma síndrome recessiva distinta.
Encontrando famílias e um quadro clínico compartilhado
A equipe vasculhou grandes projetos de sequenciamento de doenças raras e biobancos para encontrar indivíduos com duas variantes em RNU4-2 e problemas neurodesenvolvimentais. Eles identificaram, ao final, 38 pessoas de 28 famílias e reuniram dados clínicos detalhados de 31 delas. Quase todos apresentavam atraso global do desenvolvimento ou deficiência intelectual, frequentemente em graus moderado a grave, com atraso marcado na fala; cerca de metade não tinha linguagem oral funcional. Muitas crianças começaram a andar tardiamente, e mais de um quarto nunca andou independentemente aos cinco anos. Tônus muscular baixo na infância, convulsões, problemas de movimento e coordenação, e desafios comportamentais como agressividade ou traços obsessivos foram comuns. Alterações cutâneas, problemas oculares, baixa estatura e anomalias genitais em indivíduos do sexo masculino surgiram em uma minoria substancial, formando um quadro multissistêmico em vez de uma doença restrita apenas ao cérebro.
Exames cerebrais revelam uma assinatura marcante na substância branca
A ressonância magnética forneceu uma das pistas mais nítidas de que essa condição recessiva é distinta da síndrome ReNU dominante. Entre 27 indivíduos com exames ou laudos, 24 apresentaram anormalidades, e um neurorradiologista pediátrico revisou 13 exames em profundidade. Todo exame revisado mostrou envolvimento da substância branca, mais caracteristicamente uma expansão dos espaços preenchidos por líquido que circundam pequenos vasos sanguíneos (espaços perivasculares) na substância branca profunda e periventricular. Nos casos mais graves, esses espaços coalesceram em agrupamentos densos semelhantes a microcistos, frequentemente acompanhados de afinamento do corpo caloso e redução do cerebelo. Esse padrão dramático não havia sido descrito na síndrome ReNU e raramente foi observado em pessoas com variantes de RNU4-2 na população geral, tornando-se uma assinatura radiológica poderosa que pode motivar testes genéticos direcionados.
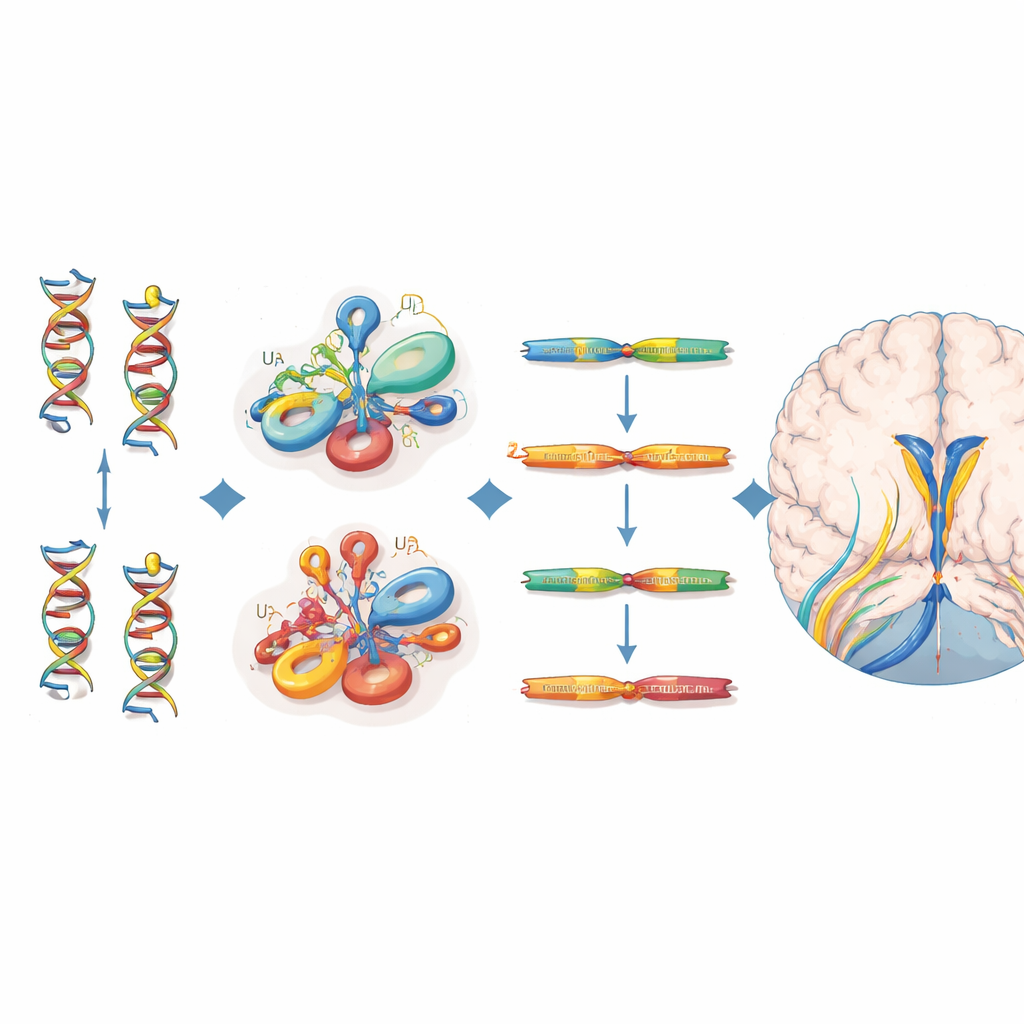
Localizando as mutações e seu impacto molecular
Todos os 38 indivíduos afetados carregavam variantes bialélicas que se agrupavam em regiões estruturais específicas do RNA U4 — particularmente a região Stem II que se liga ao RNA U6, uma curvatura acentuada chamada k-turn que recruta proteínas, e um trecho necessário para a fixação das proteínas estabilizadoras Sm. Muitas dessas posições coincidem com sítios equivalentes em um RNA relacionado, RNU4ATAC, onde mutações já são conhecidas por causar outras doenças recessivas. Para avaliar o quão nociva é cada variante, os autores se apoiaram em um experimento de edição genômica em saturação que mediu como centenas de possíveis alterações em RNU4-2 afetam a sobrevivência celular. As variantes em pacientes com doença neurodesenvolvimental tenderam a ter escores de função mais baixos do que aqueles observados em voluntários saudáveis do UK Biobank, apoiando seu papel na doença, ainda que nem toda variante clinicamente importante tenha atingido um corte estrito nesse ensaio baseado em células.
Mecanismo de doença diferente da síndrome irmã dominante
Ao analisar dados de sequenciamento de RNA, os pesquisadores mostraram que crianças com a condição recessiva de RNU4-2 têm níveis marcadamente reduzidos de RNA RNU4-2, juntamente com um aumento acentuado de um RNA intimamente relacionado, RNU4-1. A razão entre RNU4-2 e RNU4-1 foi muito menor do que em milhares de outras amostras de pacientes e pôde distinguir de forma confiável os indivíduos afetados, sugerindo um mecanismo de perda de função parcialmente compensado — mas não totalmente resgatado — pela regulação para cima de RNU4-1. Em contraste, pessoas com a síndrome ReNU dominante apresentaram, na verdade, níveis aumentados de RNU4-2 e uma mudança característica no uso de certos sítios de splicing, apontando para uma perturbação molecular qualitativamente diferente. Notavelmente, apesar do papel central do U4 no spliceossomo maior, a equipe não detectou o padrão amplo de retenção de íntrons visto em distúrbios relacionados do spliceossomo menor, o que implica um defeito mais sutil ou específico de tecido no processamento de RNA.
O que isso significa para famílias e futuros diagnósticos
Para não especialistas, a mensagem principal é que este estudo define um novo distúrbio cerebral herdado causado por ter duas cópias defeituosas de um pequeno gene de RNA não codificante. Crianças com essa condição tipicamente apresentam atraso de desenvolvimento significativo, convulsões frequentes e um padrão característico de alterações na substância branca na ressonância magnética, mas são clinicamente distintas de pacientes com a síndrome ReNU mais comum, de ação dominante. A descoberta amplia o espectro conhecido de doenças ligadas aos componentes de RNA do spliceossomo, ressalta a importância de incluir genes de pequeno RNA em painéis de testes genéticos e aponta para biomarcadores simples — como a razão de expressão RNU4-2 para RNU4-1 e a presença de dilatação perivascular extrema — que podem ajudar a encerrar a odisseia diagnóstica para famílias afetadas.
Citação: Rius, R., Blakes, A.J.M., Chen, Y. et al. Biallelic variants in the noncoding RNA gene RNU4-2 cause a recessive neurodevelopmental syndrome with distinct white matter changes. Nat Genet 58, 761–773 (2026). https://doi.org/10.1038/s41588-026-02554-6
Palavras-chave: transtorno do neurodesenvolvimento, spliceossomo, RNA não codificante, alterações da substância branca, RNU4-2