Clear Sky Science · sv
KIMMDY: en biomolekylär reaktionsemulator
Att se molekyler bilda och bryta bindningar
Livets processer bygger på otaliga små kemiska reaktioner inne i våra celler, men att verkligen i detalj se dessa reaktioner förlöpa är nästan omöjligt. Experiment visar endast suddiga medelvärden, och datorbaserade modeller har svårt att följa både rörelsen hos stora biomolekyler och bildandet och brytandet av kemiska bindningar över långa tidsskalor. Denna artikel introducerar KIMMDY, en ny typ av simulator som inte försöker spåra varje kvantdetalj i en reaktion, utan istället emulerar hur reaktioner skulle utvecklas i komplexa biologiska miljöer. Den öppnar ett fönster mot hur vävnader åldras under stress, hur DNA skadas av ljus och hur konkurrerande reaktionsvägar formar biologin.
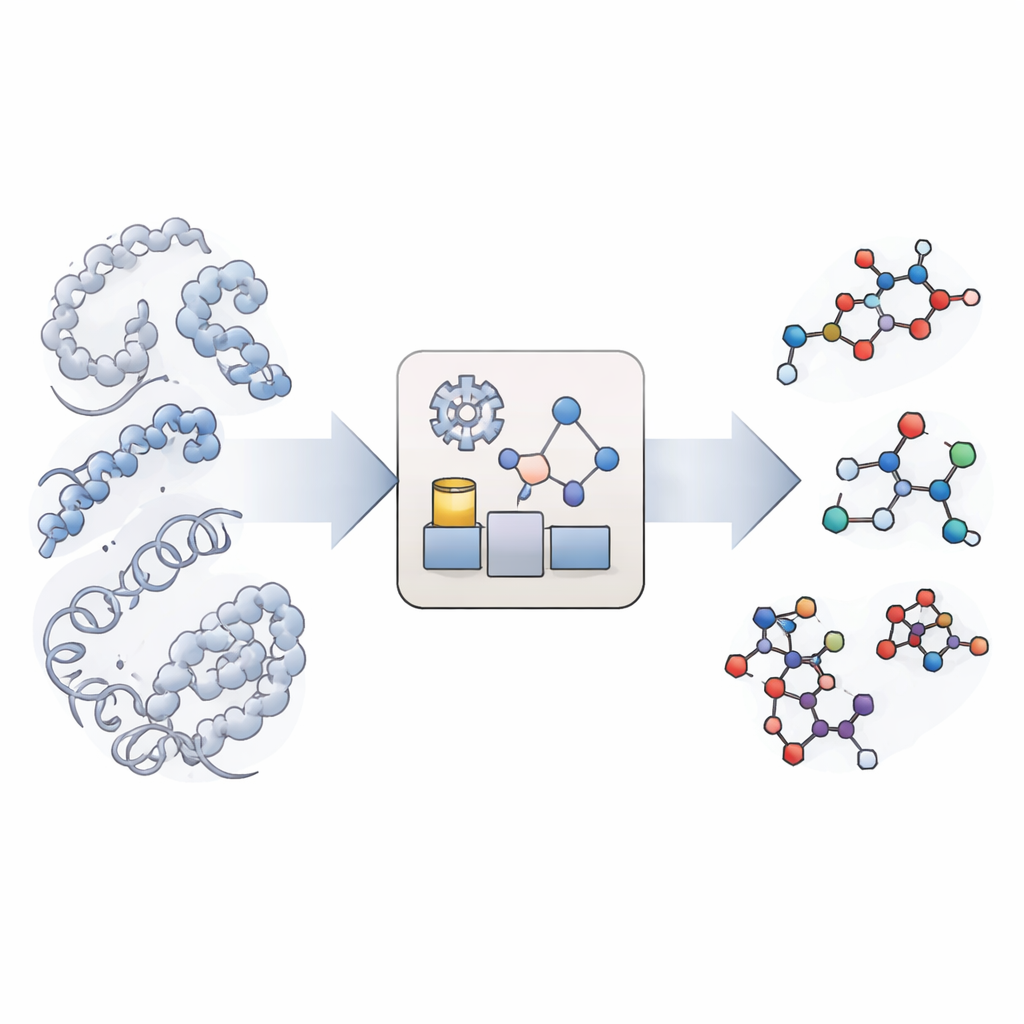
En ny metod att efterlikna kemiska reaktioner
Traditionella molekylsimuleringar är utmärkta på att visa hur stora biomolekyler som proteiner och DNA rör sig och veckas, men de behandlar kemiska bindningar som obrytbara. Verkligt reaktiva simuleringar finns, men de är så beräkningskrävande att de sällan når de långa tidsskalor som är relevanta för biologin, särskilt när många olika reaktionstyper och platser är inblandade. KIMMDY (kort för Kinetic Monte Carlo Molecular Dynamics) tar en annan väg. Den låter konventionella simuleringar hantera molekylär rörelse, och använder sedan en separat modul för att söka efter platser där reaktioner kan inträffa, uppskatta hur snabbt de skulle ske och slumpmässigt välja vilken reaktion som sker härnäst. Genom att upprepa cykler av rörelse, reaktionssökning och reaktionsval kan KIMMDY följa komplexa reaktionskaskader över tider från bråkdelar av en sekund till mycket längre, utan att någonsin beräkna en fullständig kvantmekanisk reaktionsbana.
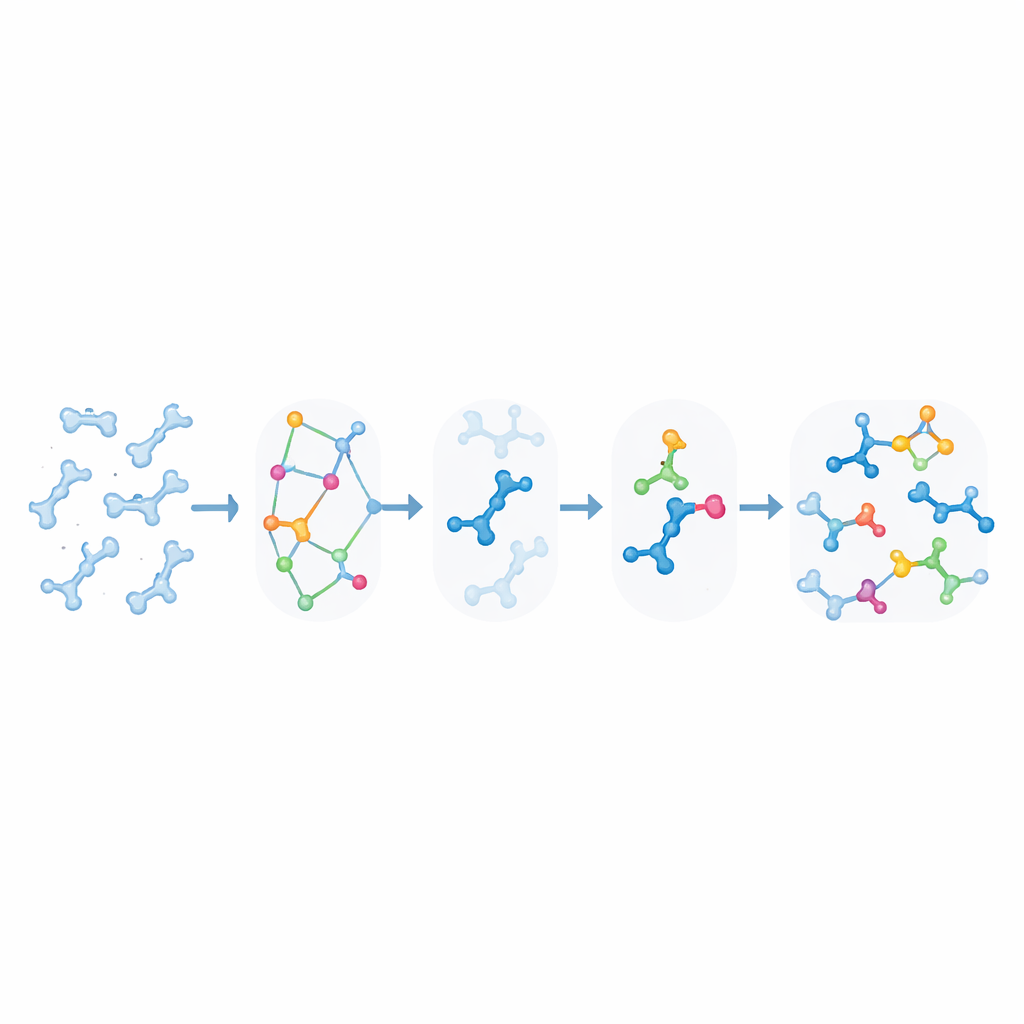
Hur reaktionsemulatorn fungerar
I hjärtat av KIMMDY finns en "reaktionsemulator" som förutsäger reaktionshastigheter utifrån molekylernas ögonblickliga former. För varje ögonblicksbild från en molekylär dynamik-körning identifierar KIMMDY möjliga reaktiva partner och använder olika modeller för att uppskatta hur stor barriären är för varje reaktion. I många fall använder den grafneuronätverk—maskininlärningsmodeller som behandlar atomer som noder och bindningar som länkar—för att gissa barriärhöjderna baserat på tidigare avancerade kvantberäkningar. I andra fall faller den tillbaka på enklare fysikbaserade formler eller heuristiska regler hämtade från experiment. Från dessa barriärer beräknas hastigheter som matas in i ett kinetiskt Monte Carlo-steg som slumpmässigt väljer nästa reaktion, avancerar den simulerade klockan och sedan uppdaterar den molekylära strukturen och kraftfältet före nästa omgång molekylrörelse.
Att följa radikalskador i proteinstommar
För att kontrollera att KIMMDY fungerar som avsett tillämpade författarna det först på en enkel klass av reaktioner i små organiska radikaler där experimentella data finns tillgängliga. Emulatorn reproducerade vilka väteförflyttningsreaktioner som är mest sannolika, även om den underskattade deras absoluta hastigheter. De gick sedan vidare till ett mycket mer komplext fall: kollagen, det fibrösa proteinet som ger bindväv dess styrka. Under mekanisk belastning kan bindningar i kollagen splittras homolytiskt och skapa mycket reaktiva radikaler som hoppar från en plats till en annan genom att överföra väteatomer längs en kedja. I en kollagenfibrill bestående av 2,6 miljoner atomer spårade KIMMDY hundratals sådana hopp och visade att radikaler kan vandra många nanometer bort från sitt ursprung. Simuleringarna avslöjade att en ovanlig korslänk (kallad PYD) och en modifierad aminosyra (DOPA) båda fungerar som exceptionellt bra radikalfällor, en slutsats som stöds av termodynamiska beräkningar och av en omtolkning av tidigare elektronspinnresonansspektra.
När olika reaktioner konkurrerar
Många biologiska system kan bryta samma bindning på mer än ett sätt, vilket leder antingen till radikalbildning eller till mer konventionella "sluten-skal"-produkter. I kollagen kan till exempel en peptidbindning antingen splittras symmetriskt till två radikaler (homolys) eller reagera med vatten och dela ojämnt (hydrolys). Direkta kvantsimuleringar av båda alternativen i en realistisk miljö skulle vara orimligt kostsamma. Med KIMMDY kombinerade författarna en fysisk modell för kraftassisterad homolys med en heuristisk, experimentbaserad modell för hydrolys och lät de två reaktionerna konkurrera både i en enskild peptid och i en tät, korslänkad kollagenfibrill. De fann att i en isolerad kedja under dragning dominerar hydrolys tydligt. I en trång, korslänkad fibrill koncentreras däremot mekanisk stress i vissa regioner så starkt att homolys blir konkurrenskraftig eller till och med snabbare, vilket förklarar varför radikalbildning observeras i verkliga vävnader.
Ljusdriven skada och korslänkar i DNA
KIMMDY belyser också hur ultraviolett ljus förändrar DNA. När två intilliggande tymidiner absorberar UV kan de slå ihop till en cyklobutan-dimer, en lesion starkt kopplad till hudcancer men också utnyttjad för att korslänka strängar i DNA-nanoteknik. Med en enkel geometrisk regel som relaterar avstånd och vinkel mellan reaktiva bindningar till reaktionssannolikhet, anpassad för att matcha småmolekylsexperiment, användes KIMMDY för att uppskatta "kvantutbyten"—hur ofta en dimer bildas per absorberad foton—för olika DNA-motiv. Emulatorn förutsade att vissa arrangemang som ofta används i DNA-origami, såsom överhäng och korsningar, faktiskt har förvånansvärt låga utbyten jämfört med standard dubbelsträngat eller nött DNA. Den antydde också att bildandet av en dimer inte dramatiskt förändrar sannolikheten för att en annan bildas i närheten, vilket innebär att tidigare skador inte nödvändigtvis förutsätter ytterligare dimerbildning.
Varför detta betyder något för biologi och design
Enkelt uttryckt erbjuder KIMMDY ett snabbt, flexibelt sätt att spela upp "tänk om"-scenarier för kemi i levande material. Genom att emulera snarare än fullt simulera reaktioner kan metoden hantera enorma system och långa tidsskalor samtidigt som den tar hänsyn till hur molekylernas ständigt föränderliga former påverkar vilka reaktioner som sker var och när. Metoden hjälpte till att upptäcka en tidigare förbisedd radikalstabiliserande plats i kollagen, klargjorde hur mekaniska krafter förskjuter balansen mellan bindningsbrytningsvägar och väckte nya frågor om hur och var DNA-korslänkar bildas under ljus. När emulatorn utökas till fler reaktionstyper och mer precisa hastighetsmodeller lovar den att bli ett kraftfullt verktyg för att förstå hur biomolekyler åldras, fungerar dåligt eller kan konstrueras för medicin och nanoteknik.
Citering: Hartmann, E., Buhr, J., Riedmiller, K. et al. KIMMDY: a biomolecular reaction emulator. Nat Commun 17, 3500 (2026). https://doi.org/10.1038/s41467-026-71955-2
Nyckelord: biomolekylära simuleringar, reaktionskinetik, molekylmodellering, DNA-skador, mekanokemi