Clear Sky Science · pl
KIMMDY: emulator reakcji biomolekularnych
Obserwowanie, jak cząsteczki tworzą i zrywają wiązania
Życie zależy od niezliczonych, drobnych reakcji chemicznych zachodzących w naszych komórkach, ale rzeczywiste obserwowanie tych reakcji w szczegółach jest niemal niemożliwe. Eksperymenty rejestrują jedynie rozmyte średnie, a modele komputerowe mają trudności, by jednocześnie śledzić ruch dużych cząsteczek biologicznych i tworzenie oraz zrywanie wiązań chemicznych w długich skalach czasowych. Ten artykuł przedstawia KIMMDY — nowy rodzaj symulatora, który nie próbuje śledzić wszystkich kwantowych detalów reakcji, lecz emuluje, jak reakcje przebiegałyby w złożonych środowiskach biologicznych. Otwiera on okno na to, jak tkanki starzeją się pod wpływem naprężeń, jak DNA ulega uszkodzeniom pod wpływem światła i jak konkurencyjne toru reakcji kształtują procesy biologiczne.
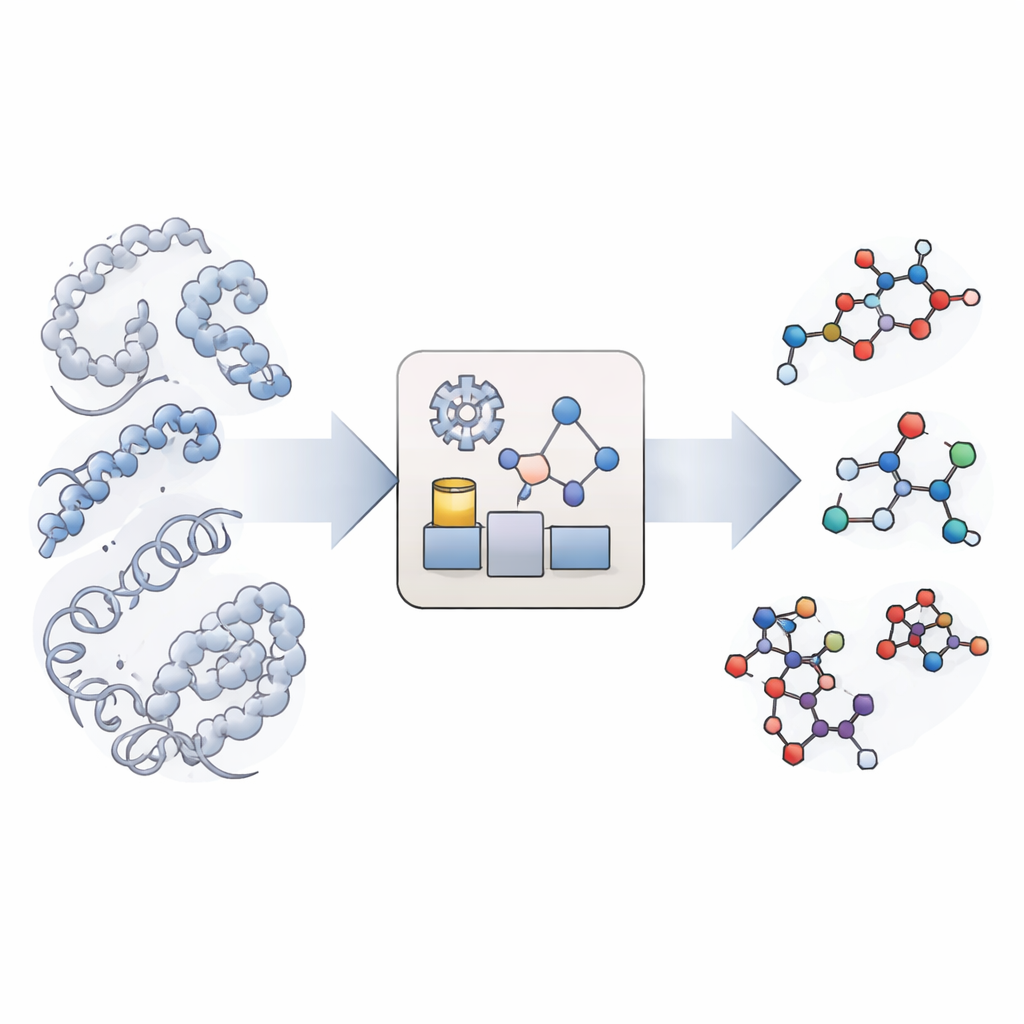
Nowy sposób na naśladowanie reakcji chemicznych
Tradycyjne symulacje molekularne świetnie pokazują, jak poruszają się i fałdują wielkie biomolekuły, takie jak białka i DNA, ale traktują wiązania chemiczne jako niełamliwe. Istnieją symulacje uwzględniające pełną reakcję, jednak są one tak kosztowne obliczeniowo, że rzadko osiągają długie skale czasowe istotne biologicznie, zwłaszcza gdy zaangażowane są rozmaite typy reakcji i miejsca reakcyjne. KIMMDY (skrót od Kinetic Monte Carlo Molecular Dynamics) wybiera inną drogę. Pozwala konwencjonalnym symulacjom zajmować się ruchem molekuł, a dodatkowy moduł wyszukuje miejsca, w których mogłyby zajść reakcje, szacuje ich szybkość i losowo wybiera, która reakcja zajdzie następna. Poprzez wielokrotne cykle ruchu, wyszukiwania reakcji i wyboru reakcji, KIMMDY może śledzić złożone kaskady reakcyjne w przedziałach czasowych od ułamków sekundy do znacznie dłuższych, bez konieczności obliczania pełnej, kwantowej ścieżki reakcyjnej.
Jak działa emulator reakcji
W sercu KIMMDY znajduje się „emulator reakcji”, który przewiduje szybkości reakcji na podstawie bieżących kształtów cząsteczek. Dla każdego zrzutu (snapshotu) z przebiegu dynamiki molekularnej KIMMDY identyfikuje możliwych partnerów reakcyjnych i stosuje różne modele do oszacowania wysokości bariery dla każdej reakcji. W wielu przypadkach wykorzystuje sieci grafowe (graph neural networks) — modele uczenia maszynowego traktujące atomy jak węzły, a wiązania jako krawędzie — aby zgadnąć wysokości barier na podstawie wcześniejszych wysokopoziomowych obliczeń kwantowych. W innych sytuacjach stosuje prostsze formuły fizyczne lub reguły heurystyczne oparte na eksperymentach. Z tych barier oblicza szybkości i przekazuje je do kroku kinetycznego Monte Carlo, który losowo wybiera kolejną reakcję, przesuwa symulowany zegar, a następnie aktualizuje strukturę molekularną i pole sił przed następną rundą ruchu molekularnego.
Śledzenie uszkodzeń rodnikowych w szkielecie białkowym
Aby sprawdzić, że KIMMDY działa zgodnie z zamierzeniem, autorzy najpierw zastosowali go do prostej klasy reakcji w małych rodnikach organicznych, dla których dostępne są dane eksperymentalne. Emulator odtworzył, które przemieszczenia atomu wodoru są najbardziej prawdopodobne, choć zwykle zaniżał bezwzględne prędkości tych reakcji. Następnie przeszli do znacznie bardziej złożonego przypadku: kolagenu, włóknistego białka nadającego tkankom łącznym wytrzymałość. Pod wpływem naprężenia mechanicznego wiązania w kolagenie mogą pękać homolitycznie, tworząc wysoce reaktywne rodniki, które przeskakują między miejscami, przekazując atomy wodoru wzdłuż łańcucha. W fibrylach kolagenu zawierających 2,6 miliona atomów KIMMDY prześledził setki takich przeskoków i wykazał, że rodniki mogą migrować wiele nanometrów od miejsca powstania. Symulacje ujawniły, że nietypowe wiązanie między łańcuchami (zwane PYD) oraz zmodyfikowany aminokwas (DOPA) działają jako wyjątkowo skuteczne pułapki na rodniki — wniosek potwierdzony obliczeniami termodynamicznymi i reinterpretacją wcześniejszych widm rezonansu spinów elektronowych.
Kiedy różne reakcje konkurują
W wielu systemach biologicznych to samo wiązanie może ulec rozerwaniu na więcej niż jeden sposób, prowadząc albo do powstania rodników, albo do bardziej konwencjonalnych produktów „zamknięto-shellowych”. W kolagenie, na przykład, wiązanie peptydowe może albo rozdzielić się symetrycznie na dwa rodniki (homoliza), albo zareagować z wodą i rozdzielić się nierównomiernie (hydroliza). Bezpośrednie kwantowe symulacje obu opcji w realistycznym środowisku byłyby niepraktyczne. Dzięki KIMMDY autorzy połączyli model fizyczny opisujący siłowo wspomaganą homolizę z heurystycznym, opartym na eksperymentach modelem hydrolizy i pozwolili obu reakcjom konkurować zarówno w pojedynczym peptydzie, jak i w gęstym włóknie kolagenu. Odkryli, że w izolowanym łańcuchu pod rozciąganiem hydroliza wyraźnie dominuje. W zatłoczonym, poprzecznie usieciowanym włóknie jednak naprężenia mechaniczne koncentrują się w określonych regionach tak silnie, że homoliza staje się konkurencyjna, a nawet szybsza, co wyjaśnia, dlaczego tworzenie się rodników obserwuje się w rzeczywistych tkankach.
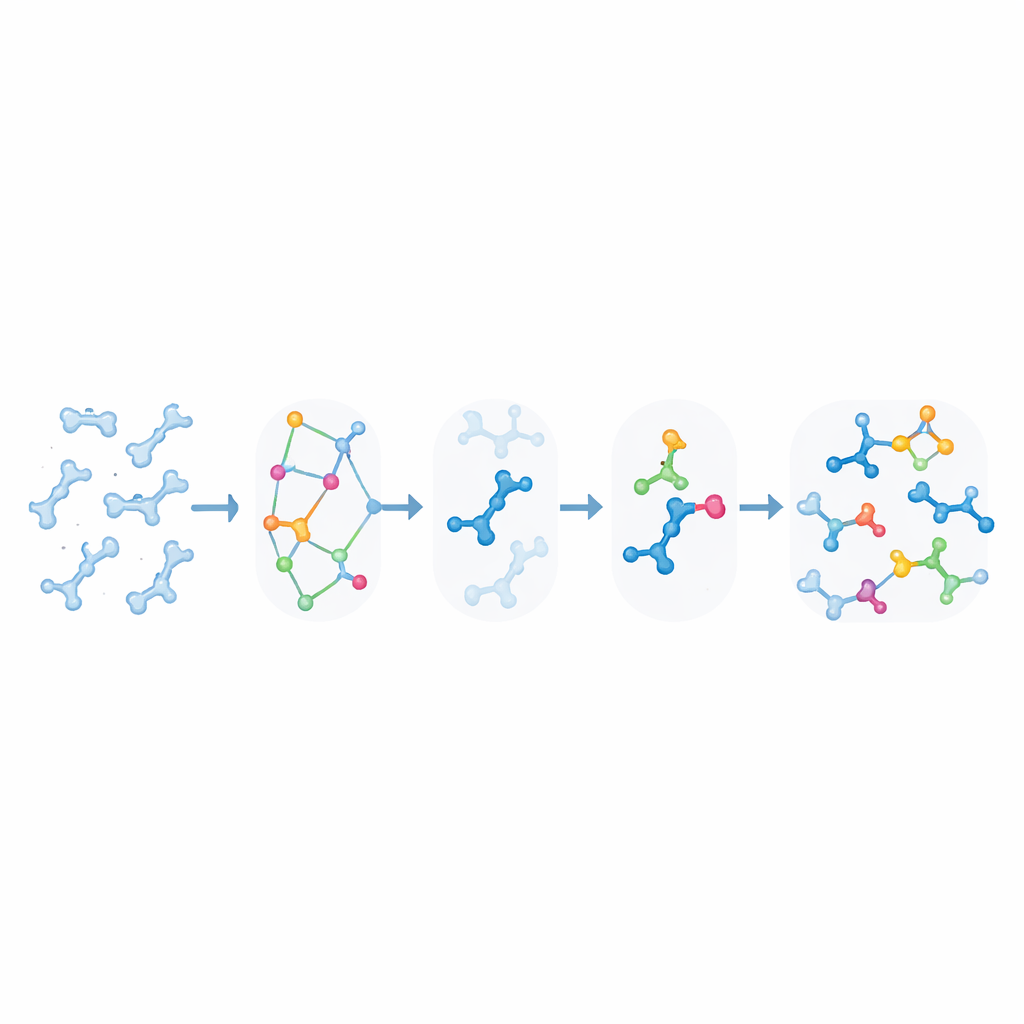
Uszkodzenia DNA wywoływane światłem i sieciowania
KIMMDY wyjaśnia również, jak ultrafiolet wpływa na DNA. Gdy dwie sąsiadujące zasady tyminy pochłoną UV, mogą zespolić się w dimery cyklobutanowe, uszkodzenia silnie związane z rakiem skóry, ale także wykorzystywane do sieciowania nici w nanotechnologii DNA. Używając prostej reguły geometrycznej wiążącej odległość i kąt między reaktywnymi wiązaniami z prawdopodobieństwem reakcji — dostrojonej do wyników eksperymentów na małych cząsteczkach — autorzy zastosowali KIMMDY do oszacowania „wydajności kwantowej” — jak często dimer powstaje na jedną pochłoniętą foton — dla różnych motywów DNA. Emulator przewidział, że niektóre układy powszechnie używane w origami DNA, takie jak wystające końcówki i skrzyżowania, mają w rzeczywistości zaskakująco niskie wydajności w porównaniu ze standardowym podwójnym heliksem lub DNA z nacięciem. Sugerował też, że powstanie jednego dimeru nie zmienia dramatycznie szans na powstanie kolejnego w pobliżu, co oznacza, że wcześniejsze uszkodzenie niekoniecznie predysponuje DNA do dalszego tworzenia dimerów.
Dlaczego to ma znaczenie dla biologii i projektowania
Mówiąc prosto, KIMMDY oferuje szybki, elastyczny sposób na przeprowadzanie scenariuszy „co jeśli” dla chemii w materii żywej. Emulując zamiast w pełni symulować reakcje, radzi sobie z ogromnymi systemami i długimi skalami czasowymi, jednocześnie uwzględniając, jak zmieniające się kształty cząsteczek wpływają na to, które reakcje zachodzą, gdzie i kiedy. Metoda pomogła odkryć wcześniej przeoczony stabilizujący rodnik miejsce w kolagenie, wyjaśniła, jak siły mechaniczne przesuwają równowagę między ścieżkami pękania wiązań i postawiła nowe pytania o to, gdzie i jak powstają skrzyżowania w DNA pod wpływem światła. W miarę rozszerzania emulatora o więcej typów reakcji i dokładniejszych modeli szybkości, ma on szansę stać się potężnym narzędziem do rozumienia, jak biomolekuły się starzeją, zawodzą lub mogą być projektowane dla medycyny i nanotechnologii.
Cytowanie: Hartmann, E., Buhr, J., Riedmiller, K. et al. KIMMDY: a biomolecular reaction emulator. Nat Commun 17, 3500 (2026). https://doi.org/10.1038/s41467-026-71955-2
Słowa kluczowe: symulacje biomolekularne, kinetyka reakcji, modelowanie molekularne, uszkodzenia DNA, mechanochemia