Clear Sky Science · es
KIMMDY: un emulador de reacciones biomoleculares
Observando cómo las moléculas forman y rompen enlaces
La vida depende de innumerables reacciones químicas diminutas dentro de nuestras células, pero observar esas reacciones desarrollarse en detalle es casi imposible. Los experimentos solo muestran promedios borrosos, y los modelos computacionales tienen dificultades para seguir tanto el movimiento de grandes moléculas biológicas como la formación y ruptura de enlaces químicos en escalas de tiempo largas. Este artículo presenta KIMMDY, un nuevo tipo de simulador que no intenta rastrear cada detalle cuántico de una reacción, sino que emula cómo se desarrollarían las reacciones en entornos biológicos complejos. Abre una ventana sobre cómo los tejidos envejecen bajo estrés, cómo la luz daña el ADN y cómo las vías reactivas en competencia dan forma a la biología.
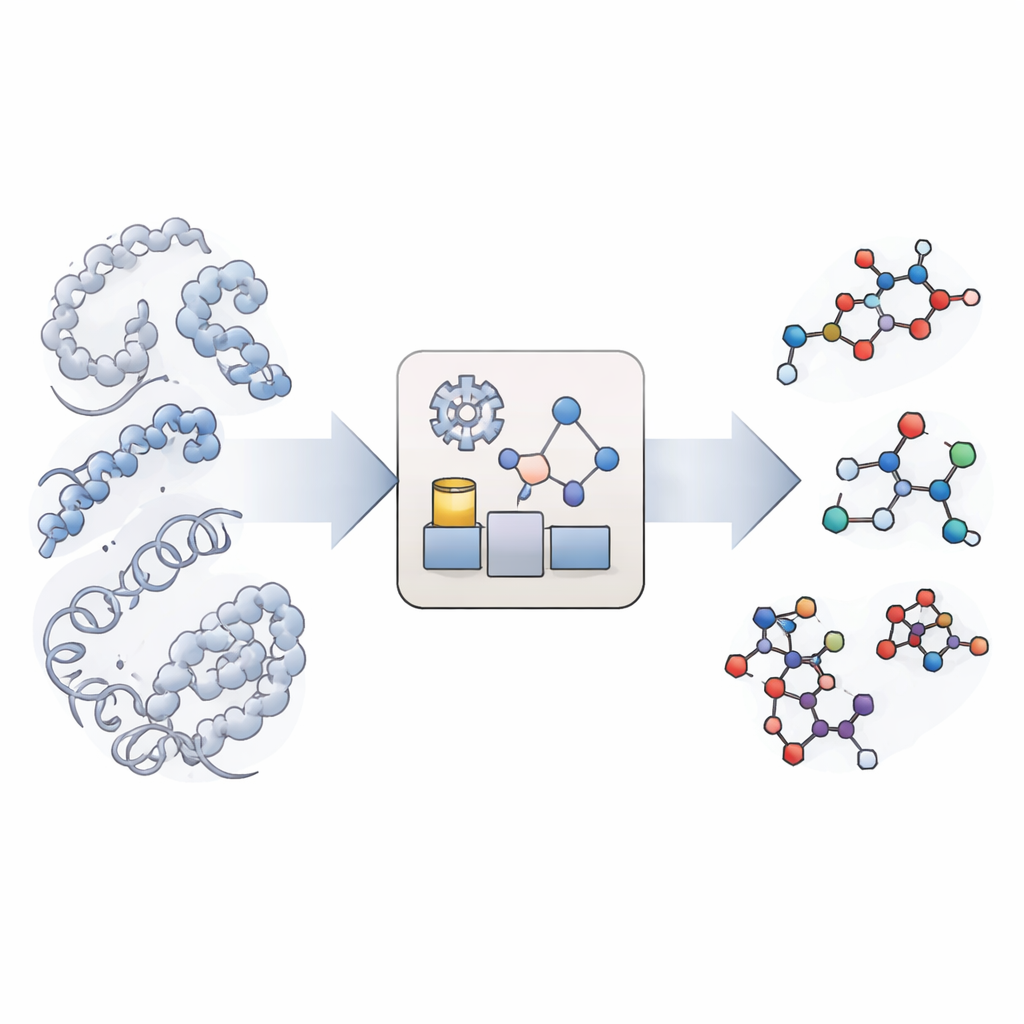
Una nueva forma de imitar reacciones químicas
Las simulaciones moleculares tradicionales son excelentes para mostrar cómo se mueven y pliegan biomoléculas grandes como proteínas y ADN, pero tratan los enlaces químicos como irrompibles. Existen simulaciones verdaderamente reactivas, sin embargo son tan exigentes computacionalmente que rara vez alcanzan las escalas de tiempo relevantes para la biología, especialmente cuando intervienen muchos tipos de reacción y sitios distintos. KIMMDY (siglas de Kinetic Monte Carlo Molecular Dynamics) toma otra ruta. Permite que las simulaciones convencionales manejen el movimiento molecular y, a continuación, utiliza un módulo adicional para buscar lugares donde podrían ocurrir reacciones, estimar qué velocidad tendrían y elegir aleatoriamente qué reacción ocurre a continuación. Al recorrer repetidamente movimiento, búsqueda de reacciones y elección de reacción, KIMMDY puede seguir cascadas reactivas complejas en tiempos que van desde fracciones de segundo hasta mucho más, sin calcular nunca una vía de reacción completa a nivel cuántico.
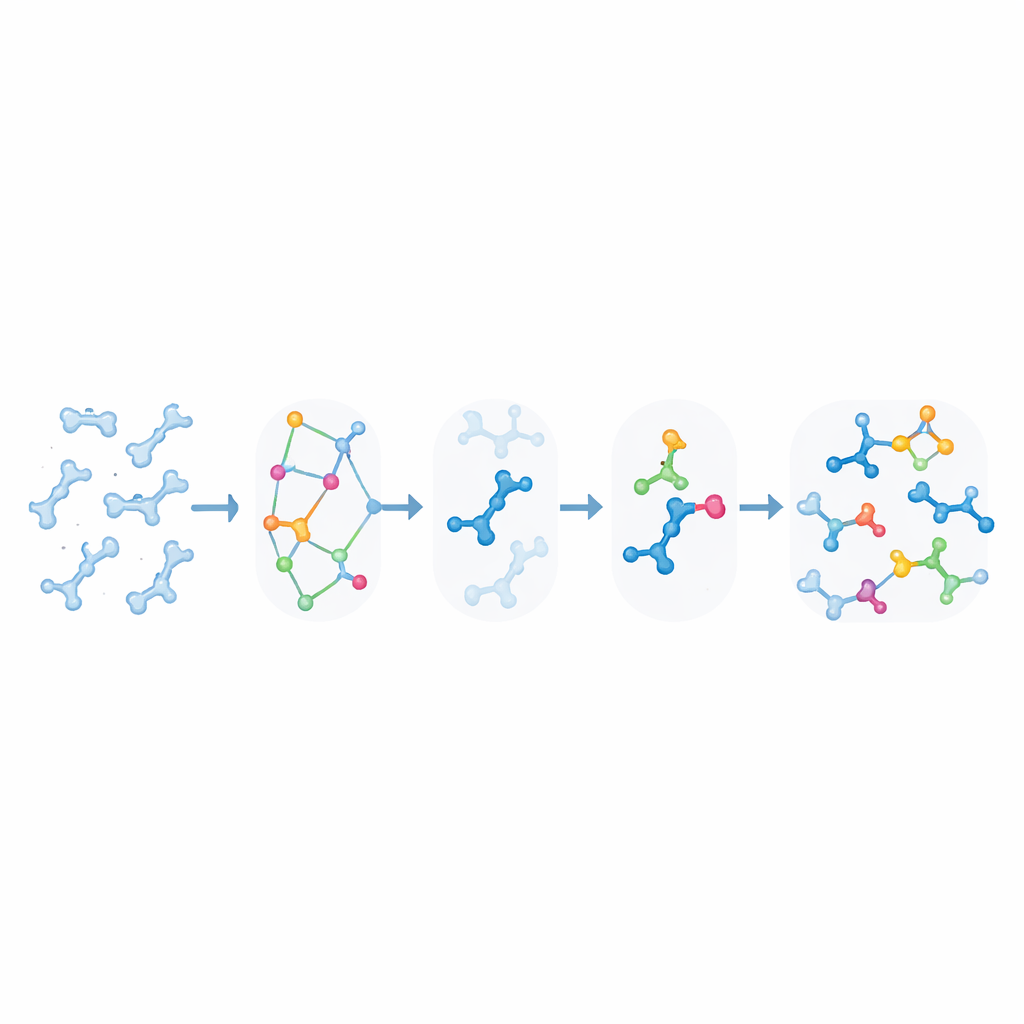
Cómo funciona el emulador de reacciones
En el núcleo de KIMMDY hay un “emulador de reacciones” que predice tasas de reacción a partir de las formas instantáneas de las moléculas. Para cada instantánea de una ejecución de dinámica molecular, KIMMDY identifica posibles parejas reactivas y emplea distintos modelos para estimar cuán alta es la barrera para cada reacción. En muchos casos utiliza redes neuronales gráficas—modelos de aprendizaje automático que tratan los átomos como nodos y los enlaces como aristas—para adivinar las alturas de las barreras basándose en cálculos cuánticos previos de alto nivel. En otros, recurre a fórmulas físicas más simples o a reglas heurísticas extraídas de experimentos. A partir de esas barreras calcula tasas y las introduce en un paso de Monte Carlo cinético que elige aleatoriamente la siguiente reacción, adelanta el reloj simulado y luego actualiza la estructura molecular y el campo de fuerzas antes de la siguiente ronda de movimiento molecular.
Siguiendo el daño radical en andamios proteicos
Para comprobar que KIMMDY funciona como se espera, los autores lo aplicaron primero a una clase simple de reacciones en pequeños radicales orgánicos donde hay datos experimentales disponibles. El emulador reprodujo qué reacciones de desplazamiento de hidrógeno son más probables, aunque subestimó sus velocidades absolutas. Luego abordaron un caso mucho más complejo: el colágeno, la proteína fibrosa que da resistencia a los tejidos conectivos. Bajo estrés mecánico, los enlaces en el colágeno pueden partirse homolíticamente, creando radicales altamente reactivos que saltan de un sitio a otro pasando átomos de hidrógeno a lo largo de una cadena. En un fibrilar de colágeno compuesto por 2,6 millones de átomos, KIMMDY rastreó cientos de estos saltos y mostró que los radicales pueden migrar muchos nanómetros lejos de su origen. Las simulaciones revelaron que un entrecruzamiento inusual (llamado PYD) y un aminoácido modificado (DOPA) actúan ambos como trampas de radicales excepcionalmente eficaces, una conclusión respaldada por cálculos termodinámicos y por reinterpretar espectros previos de resonancia paramagnética electrónica.
Cuando diferentes reacciones compiten
Muchos sistemas biológicos pueden romper el mismo enlace de más de una manera, dando lugar ya sea a la formación de radicales o a productos más convencionales de “capa cerrada”. En el colágeno, por ejemplo, un enlace peptídico puede partirse simétricamente en dos radicales (homólisis) o reaccionar con agua y partirse de forma desigual (hidrólisis). Simulaciones cuánticas directas de ambas opciones en un entorno realista serían prohibitivas. Con KIMMDY, los autores combinaron un modelo físico para la homólisis asistida por fuerza con un modelo heurístico, basado en experimentos, para la hidrólisis y dejaron que ambas reacciones compitieran dentro de una única péptida y de un fibrilar de colágeno denso. Encontraron que en una cadena aislada bajo tracción, la hidrólisis domina claramente. Sin embargo, en un fibrilar concurrido y entrecruzado, el estrés mecánico se concentra en ciertas regiones con tanta intensidad que la homólisis se vuelve competitiva o incluso más rápida, explicando por qué se observa formación de radicales en tejidos reales.
Daño inducido por la luz y entrecruzamientos en el ADN
KIMMDY también aclara cómo la luz ultravioleta altera el ADN. Cuando dos timinas vecinas absorben UV, pueden fusionarse en un dímero de ciclobutano, una lesión fuertemente relacionada con el cáncer de piel pero también aprovechada para entrecruzar hebras en nanotecnia de ADN. Usando una regla geométrica simple que relaciona distancia y ángulo entre enlaces reactivos con la probabilidad de reacción, afinada para ajustarse a experimentos con pequeñas moléculas, los autores emplearon KIMMDY para estimar “rendimientos cuánticos”: con qué frecuencia se forma un dímero por fotón absorbido, para varios motivos de ADN. El emulador predijo que algunos arreglos comúnmente usados en ADN origami, como salientes y cruces, tienen en realidad rendimientos sorprendentemente bajos en comparación con ADN de doble hebra estándar o con roturas (nicks). También sugirió que la formación de un dímero no cambia de manera dramática las posibilidades de que se forme otro cercano, lo que implica que el daño previo no necesariamente prepara al ADN para más dímeros.
Por qué esto importa para la biología y el diseño
En términos sencillos, KIMMDY ofrece una forma rápida y flexible de representar escenarios “qué pasaría si” para la química dentro de la materia viviente. Al emular en lugar de simular completamente las reacciones, puede manejar sistemas enormes y escalas de tiempo largas a la vez que tiene en cuenta cómo las formas siempre cambiantes de las moléculas influyen en qué reacciones ocurren, dónde y cuándo. El método ayudó a descubrir un sitio estabilizador de radicales previamente pasado por alto en el colágeno, clarificó cómo las fuerzas mecánicas desplazan el equilibrio entre vías de ruptura de enlaces y planteó nuevas preguntas sobre cómo y dónde se forman entrecruzamientos en el ADN bajo la luz. A medida que el emulador se extienda a más tipos de reacciones y a modelos de tasas más precisos, promete convertirse en una herramienta poderosa para entender cómo las biomoléculas envejecen, fallan o pueden diseñarse para la medicina y la nanotecnología.
Cita: Hartmann, E., Buhr, J., Riedmiller, K. et al. KIMMDY: a biomolecular reaction emulator. Nat Commun 17, 3500 (2026). https://doi.org/10.1038/s41467-026-71955-2
Palabras clave: simulaciones biomoleculares, cinética de reacciones, modelado molecular, daño en el ADN, mecanoquímica