Clear Sky Science · de
KIMMDY: ein Emulator biomolekularer Reaktionen
Beobachten, wie Moleküle Bindungen formen und brechen
Das Leben beruht auf unzähligen winzigen chemischen Reaktionen in unseren Zellen, doch diese Reaktionen detailliert ablaufen zu sehen, ist nahezu unmöglich. Experimente liefern nur verschwommene Mittelwerte, und Computermodelle tun sich schwer damit, sowohl die Bewegung großer biologischer Moleküle als auch das Bilden und Brechen chemischer Bindungen über lange Zeiten nachzuverfolgen. Dieser Artikel stellt KIMMDY vor, einen neuen Typ von Simulator, der nicht versucht, jedes quantenmechanische Detail einer Reaktion zu verfolgen, sondern stattdessen nachahmt, wie Reaktionen in komplexen biologischen Umgebungen ablaufen würden. Er öffnet ein Fenster dafür, wie Gewebe unter Stress altern, wie DNA durch Licht geschädigt wird und wie konkurrierende Reaktionswege die Biologie formen.
Ein neuer Weg, chemische Reaktionen zu imitieren
Traditionelle molekulare Simulationen sind exzellent darin zu zeigen, wie große Biomoleküle wie Proteine und DNA sich bewegen und falten, behandeln chemische Bindungen aber als unverletzlich. Wirklich reaktive Simulationen existieren zwar, sind jedoch so rechenaufwendig, dass sie selten die für die Biologie relevanten langen Zeitskalen erreichen können, insbesondere wenn viele verschiedene Reaktionstypen und -stellen beteiligt sind. KIMMDY (Kurzform für Kinetic Monte Carlo Molecular Dynamics) geht einen anderen Weg. Es lässt konventionelle Simulationen die molekulare Bewegung übernehmen und nutzt dann ein zusätzliches Modul, um nach Stellen zu suchen, an denen Reaktionen auftreten könnten, abzuschätzen, wie schnell sie ablaufen würden, und stochastisch auszuwählen, welche Reaktion als nächstes stattfindet. Durch wiederholte Zyklen aus Bewegung, Reaktionssuche und Reaktionswahl kann KIMMDY komplexe Reaktionskaskaden über Zeiträume von Bruchteilen einer Sekunde bis zu deutlich längeren Zeiten verfolgen, ohne jemals einen vollständigen quantenmechanischen Reaktionsweg zu berechnen.
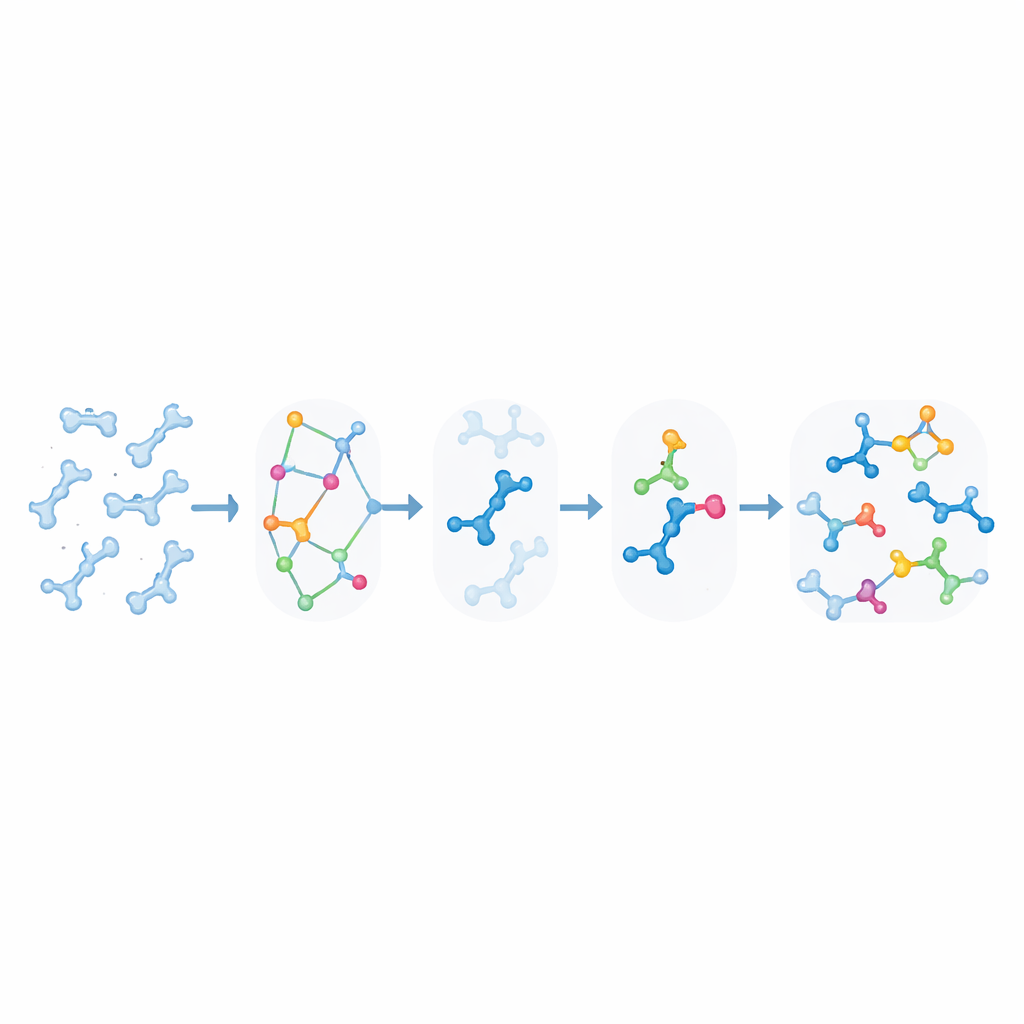
Wie der Reaktions-Emulator funktioniert
Im Zentrum von KIMMDY steht ein „Reaktions-Emulator“, der aus den momentanen Molekülgestalten Reaktionsraten vorhersagt. Für jeden Schnappschuss aus einem Molekulardynamiklauf identifiziert KIMMDY mögliche Reaktionspartner und verwendet unterschiedliche Modelle, um abzuschätzen, wie hoch die Barriere für jede Reaktion ist. In vielen Fällen setzt es Graph-Neuronale-Netzwerke ein — maschinelle Lernmodelle, die Atome als Knoten und Bindungen als Kanten behandeln — um die Barrieren basierend auf vorherigen hochrangigen Quantenberechnungen zu schätzen. In anderen Fällen greift es auf einfachere, physikbasierte Formeln oder heuristische Regeln aus Experimenten zurück. Aus diesen Barrieren berechnet es Raten und speist sie in einen kinetischen Monte-Carlo-Schritt ein, der zufällig die nächste Reaktion auswählt, die simulierte Uhr voranschreiten lässt und anschließend die molekulare Struktur und das Kraftfeld aktualisiert, bevor die nächste Runde molekularer Bewegung beginnt.
Radikalschäden in Proteingerüsten verfolgen
Um zu prüfen, dass KIMMDY wie beabsichtigt funktioniert, wandten die Autoren es zunächst auf eine einfache Klasse von Reaktionen in kleinen organischen Radikalen an, für die experimentelle Daten vorliegen. Der Emulator reproduzierte, welche Wasserstoff‑Verschiebe‑Reaktionen am wahrscheinlichsten sind, auch wenn er ihre absoluten Geschwindigkeiten unterschätzte. Anschließend gingen sie zu einem deutlich komplexeren Fall über: Kollagen, das faserige Protein, das Bindegeweben seine Festigkeit verleiht. Unter mechanischer Belastung können Bindungen in Kollagen homolytisch spalten und hochreaktive Radikale erzeugen, die durch Weitergabe von Wasserstoffatomen von einer Stelle zur anderen „hüpfen“. In einem Kollagenfibrill mit 2,6 Millionen Atomen verfolgte KIMMDY Hunderte dieser Hüpfer und zeigte, dass Radikale viele Nanometer von ihrem Entstehungsort wandern können. Die Simulationen enthüllten, dass eine ungewöhnliche Querbindung (als PYD bezeichnet) und eine modifizierte Aminosäure (DOPA) beide als außergewöhnlich gute Radikalfänger wirken — eine Schlussfolgerung, die durch thermodynamische Berechnungen und die Neuinterpretation früherer Elektronenspin-Resonanzspektren gestützt wird.
Wenn verschiedene Reaktionen konkurrieren
Viele biologische Systeme können dieselbe Bindung auf mehr als eine Weise aufbrechen, was entweder zur Radikalbildung oder zu konventionelleren „geschlossenen Schalen“-Produkten führt. In Kollagen kann sich zum Beispiel eine Peptidbindung entweder symmetrisch in zwei Radikale spalten (Homolyse) oder mit Wasser reagieren und unsymmetrisch spalten (Hydrolyse). Direkte Quanten‑Simulationen beider Möglichkeiten in einer realistischen Umgebung wären prohibitiv. Mit KIMMDY kombinierten die Autoren ein physikalisches Modell für kraftunterstützte Homolyse mit einem heuristischen, experimentbasierten Modell für Hydrolyse und ließen die beiden Reaktionen in einer einzelnen Peptidkette sowie in einem dichten Kollagenfibrill konkurrieren. Sie fanden heraus, dass bei einer isolierten Kette unter Zug die Hydrolyse klar dominiert. In einem dicht gepackten, quervernetzten Fibrill jedoch konzentriert sich die mechanische Spannung in bestimmten Regionen so stark, dass die Homolyse konkurrenzfähig oder sogar schneller wird — was erklärt, warum Radikalbildung in realen Geweben beobachtet wird.
Lichtgetriebene Schäden und Querbindungen in der DNA
KIMMDY beleuchtet auch, wie ultraviolettes Licht die DNA verändert. Wenn zwei benachbarte Thyminbasen UV absorbieren, können sie zu einem Cyclobutan-Dimer verschmelzen — eine Läsion, die stark mit Hautkrebs verbunden ist, aber auch zur Quervernetzung von Strängen in der DNA-Nanotechnologie genutzt wird. Mittels einer einfachen geometrischen Regel, die Abstand und Winkel zwischen reaktiven Bindungen mit der Reaktionswahrscheinlichkeit verknüpft und an Messungen mit Kleinmolekülen angepasst wurde, nutzten die Autoren KIMMDY, um „quantenausbeuten“ abzuschätzen — wie oft ein Dimer pro absorbiertem Photon bildet — für verschiedene DNA‑Motive. Der Emulator sagte voraus, dass einige Anordnungen, die häufig in DNA‑Origami verwendet werden, wie Überhänge und Kreuzungen, tatsächlich überraschend niedrige Ausbeuten im Vergleich zu standardmäßiger doppelsträngiger oder genickter DNA aufweisen. Er schlug außerdem vor, dass die Bildung eines Dimers die Wahrscheinlichkeit, ein weiteres in der Nähe zu bilden, nicht dramatisch verändert, was impliziert, dass vorausgegangener Schaden DNA nicht unbedingt für weitere Dimerbildung präpariert.
Warum das für Biologie und Design wichtig ist
Einfach gesagt bietet KIMMDY eine schnelle, flexible Möglichkeit, „Was‑wenn“-Szenarien für Chemie in lebendem Material durchzuspielen. Indem es Reaktionen emuliert statt vollständig zu simulieren, kann es enorme Systeme und lange Zeitskalen behandeln und zugleich berücksichtigen, wie die sich ständig ändernden Gestalten von Molekülen beeinflussen, welche Reaktionen wo und wann stattfinden. Die Methode half dabei, eine zuvor übersehene radikalstabilisierende Stelle in Kollagen aufzudecken, zu klären, wie mechanische Kräfte das Gleichgewicht zwischen Bindungs‑Aufbrechungswegen verschieben, und neue Fragen darüber aufzuwerfen, wie und wo DNA‑Querbindungen unter Lichteinfluss entstehen. Wenn der Emulator auf mehr Reaktionstypen und genauere Ratenmodelle ausgeweitet wird, verspricht er ein mächtiges Werkzeug zu werden, um zu verstehen, wie Biomoleküle altern, fehlfunktionieren oder für Medizin und Nanotechnologie entworfen werden können.
Zitation: Hartmann, E., Buhr, J., Riedmiller, K. et al. KIMMDY: a biomolecular reaction emulator. Nat Commun 17, 3500 (2026). https://doi.org/10.1038/s41467-026-71955-2
Schlüsselwörter: biomolekulare Simulationen, Reaktionskinetik, molekulares Modellieren, DNA-Schädigung, Mechanochemie