Clear Sky Science · en
KIMMDY: a biomolecular reaction emulator
Watching Molecules Make and Break Bonds
Life depends on countless tiny chemical reactions inside our cells, but actually watching those reactions unfold in detail is almost impossible. Experiments see only blurry averages, and computer models struggle to follow both the motion of large biological molecules and the making and breaking of chemical bonds over long times. This article introduces KIMMDY, a new kind of simulator that does not try to track every quantum detail of a reaction, but instead emulates how reactions would play out in complex biological environments. It opens a window on how tissues age under stress, how DNA is damaged by light, and how competing reaction pathways shape biology.
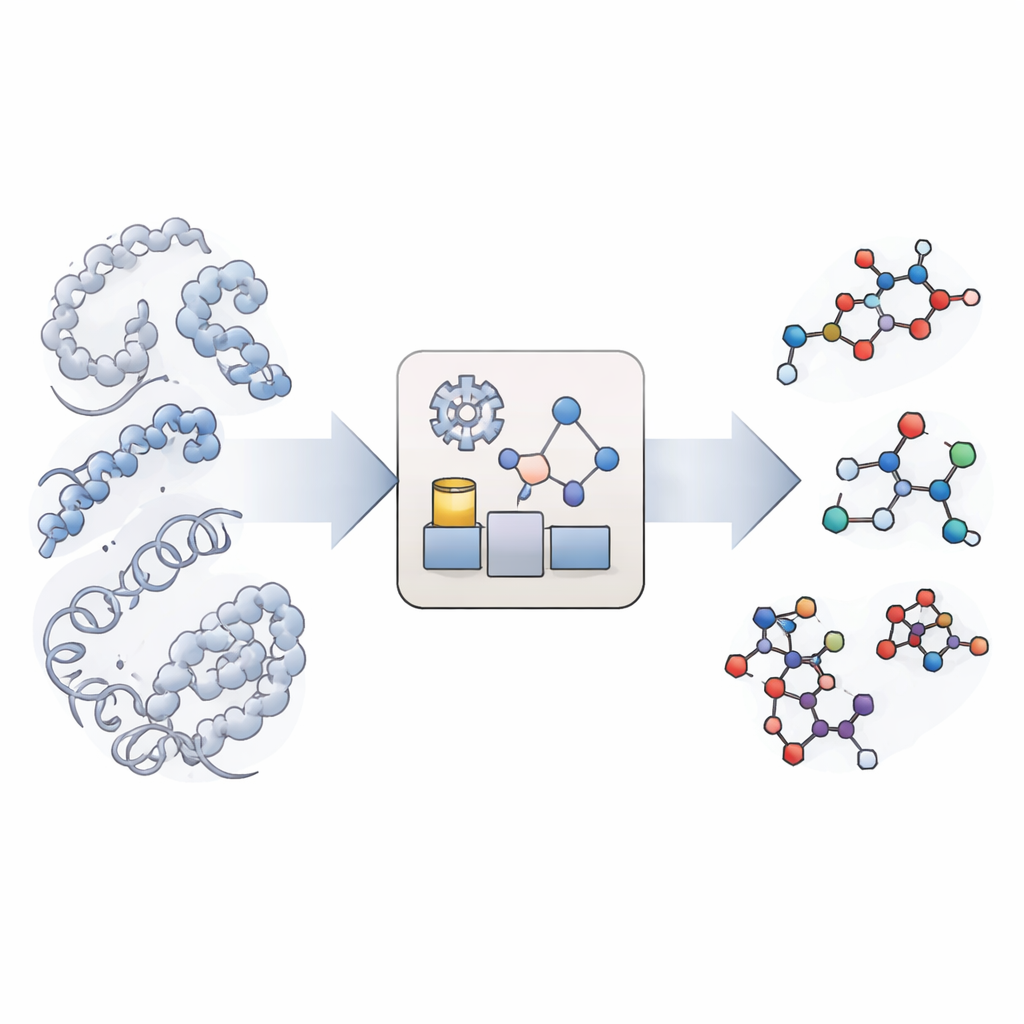
A New Way to Imitate Chemical Reactions
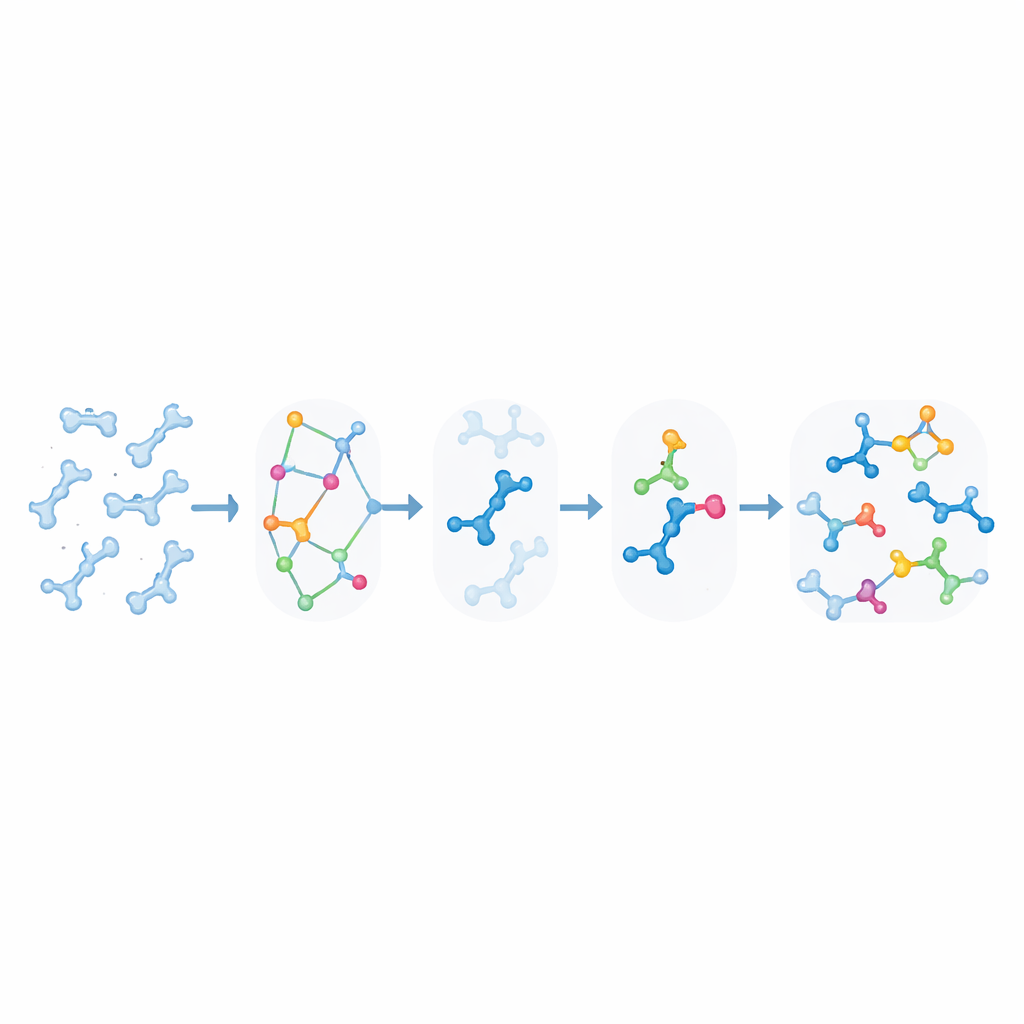
Traditional molecular simulations are excellent at showing how big biomolecules such as proteins and DNA move and fold, but they treat chemical bonds as unbreakable. Truly reactive simulations exist, yet they are so computationally demanding that they can rarely reach the long timescales relevant for biology, especially when many different reaction types and sites are involved. KIMMDY (short for Kinetic Monte Carlo Molecular Dynamics) takes a different route. It lets conventional simulations handle the molecular motion, then uses an additional module to search for places where reactions could occur, estimate how fast they would happen, and randomly choose which reaction takes place next. By repeatedly cycling through motion, reaction search, and reaction choice, KIMMDY can follow complex reaction cascades over times ranging from fractions of a second to much longer, without ever computing a full quantum-level reaction pathway.
How the Reaction Emulator Works
At the heart of KIMMDY is a "reaction emulator" that predicts reaction rates from the instantaneous shapes of molecules. For each snapshot from a molecular dynamics run, KIMMDY identifies possible reacting partners and uses different models to estimate how large the barrier is for each reaction. In many cases, it employs graph neural networks—machine-learning models that treat atoms as nodes and bonds as links—to guess the barrier heights based on previous high-level quantum calculations. In other cases, it falls back on simpler physics-based formulas or heuristic rules drawn from experiments. From those barriers, it computes rates and feeds them into a kinetic Monte Carlo step that randomly chooses the next reaction, advances the simulated clock, and then updates the molecular structure and force field before the next round of molecular motion.
Following Radical Damage in Protein Scaffolds
To check that KIMMDY works as intended, the authors first applied it to a simple class of reactions in small organic radicals where experimental data are available. The emulator reproduced which hydrogen-shift reactions are most likely, even if it underestimated their absolute speeds. They then moved on to a much more complex case: collagen, the fibrous protein that gives connective tissues their strength. Under mechanical stress, bonds in collagen can split homolytically, creating highly reactive radicals that hop from one site to another by passing hydrogen atoms along a chain. In a collagen fibril made of 2.6 million atoms, KIMMDY traced hundreds of these hops and showed that radicals can migrate many nanometers away from their origin. The simulations revealed that an unusual crosslink (called PYD) and a modified amino acid (DOPA) both act as exceptionally good radical traps, a conclusion supported by thermodynamic calculations and by reinterpreting earlier electron spin resonance spectra.
When Different Reactions Compete
Many biological systems can break the same bond in more than one way, leading either to radical formation or to more conventional "closed-shell" products. In collagen, for instance, a peptide bond can either split symmetrically into two radicals (homolysis) or react with water and split unevenly (hydrolysis). Direct quantum simulations of both options in a realistic environment would be prohibitive. With KIMMDY, the authors combined a physical model for force-assisted homolysis with a heuristic, experiment-based model for hydrolysis and let the two reactions compete inside both a single peptide and a dense collagen fibril. They found that in an isolated chain under pulling, hydrolysis clearly dominates. In a crowded, crosslinked fibril, however, mechanical stress concentrates in certain regions so strongly that homolysis becomes competitive or even faster, explaining why radical formation is observed in real tissues.
Light-Driven Damage and Crosslinks in DNA
KIMMDY also illuminates how ultraviolet light alters DNA. When two neighboring thymine bases absorb UV, they can fuse into a cyclobutane dimer, a lesion strongly linked to skin cancer but also exploited to crosslink strands in DNA nanotechnology. Using a simple geometric rule that relates distance and angle between reactive bonds to reaction likelihood, tuned to match small-molecule experiments, the authors used KIMMDY to estimate "quantum yields"—how often a dimer forms per absorbed photon—for various DNA motifs. The emulator predicted that some arrangements commonly used in DNA origami, such as overhangs and crossovers, actually have surprisingly low yields compared with standard double-stranded or nicked DNA. It also suggested that forming one dimer does not dramatically change the chances of forming another nearby, implying that prior damage does not necessarily prime DNA for further dimers.
Why This Matters for Biology and Design
In plain terms, KIMMDY offers a fast, flexible way to play out "what if" scenarios for chemistry inside living matter. By emulating rather than fully simulating reactions, it can handle enormous systems and long timescales while still accounting for how the ever-changing shapes of molecules influence which reactions happen where and when. The method helped uncover a previously overlooked radical-stabilizing site in collagen, clarified how mechanical forces shift the balance between bond-breaking pathways, and raised new questions about how and where DNA crosslinks form under light. As the emulator is extended to more reaction types and more accurate rate models, it promises to become a powerful tool for understanding how biomolecules age, malfunction, or can be engineered for medicine and nanotechnology.
Citation: Hartmann, E., Buhr, J., Riedmiller, K. et al. KIMMDY: a biomolecular reaction emulator. Nat Commun 17, 3500 (2026). https://doi.org/10.1038/s41467-026-71955-2
Keywords: biomolecular simulations, reaction kinetics, molecular modelling, DNA damage, mechanochemistry