Clear Sky Science · fr
KIMMDY : un émulateur de réactions biomoléculaires
Observer les molécules former et rompre des liaisons
La vie repose sur d'innombrables petites réactions chimiques au sein de nos cellules, mais observer ces réactions en détail est presque impossible. Les expériences n'offrent que des moyennes floues, et les modèles informatiques peinent à suivre à la fois le mouvement de grandes molécules biologiques et la formation/rupture de liaisons chimiques sur de longues échelles de temps. Cet article présente KIMMDY, un nouveau type de simulateur qui n'essaie pas de suivre chaque détail quantique d'une réaction, mais qui émule plutôt la manière dont les réactions se dérouleraient dans des environnements biologiques complexes. Il ouvre une fenêtre sur la façon dont les tissus vieillissent sous contrainte, comment l'ADN est endommagé par la lumière et comment des voies réactionnelles concurrentes façonnent la biologie.
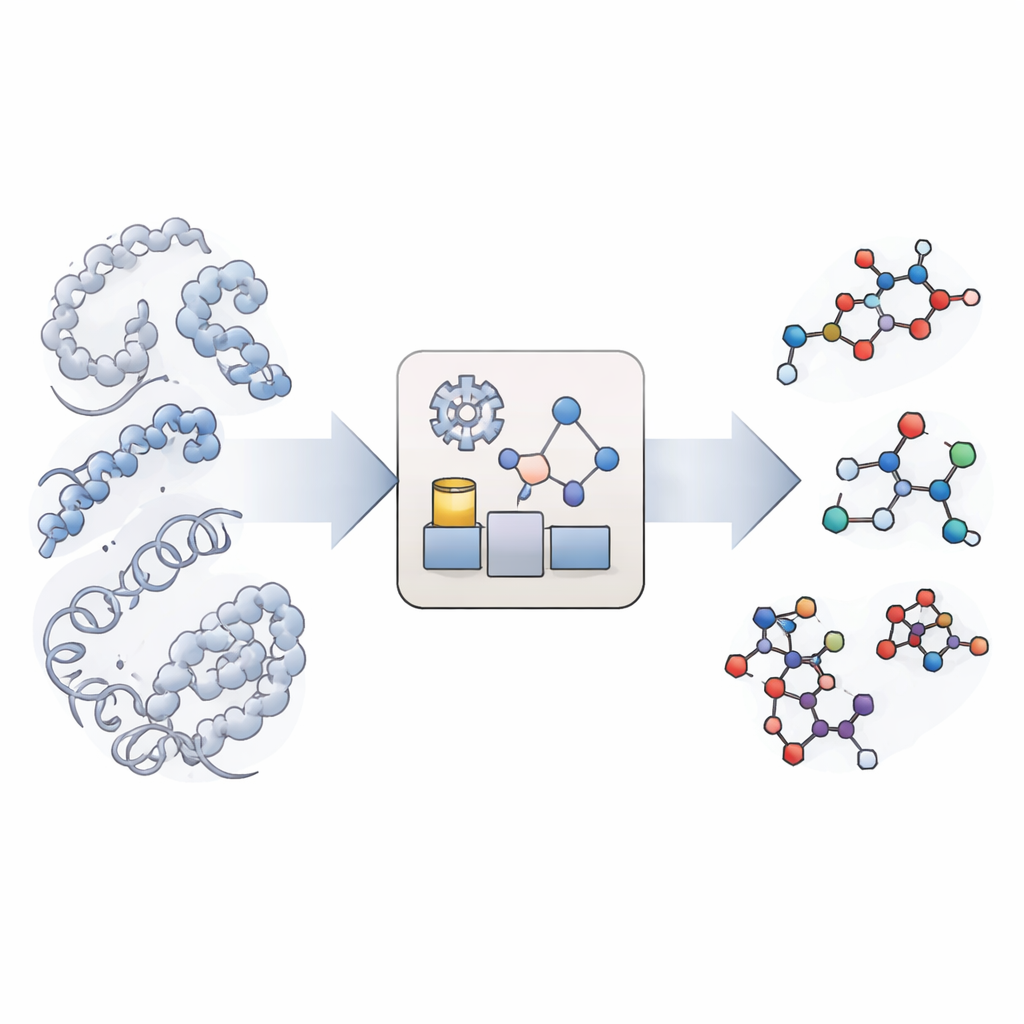
Une nouvelle façon d'imiter les réactions chimiques
Les simulations moléculaires traditionnelles excellent pour montrer comment de grandes biomolécules comme les protéines et l'ADN se déplacent et se replient, mais elles traitent les liaisons chimiques comme indestructibles. Des simulations réellement réactives existent, mais elles sont si coûteuses en calcul qu'elles atteignent rarement les échelles de temps pertinentes pour la biologie, surtout lorsqu'interviennent de nombreux types de réactions et sites distincts. KIMMDY (pour Kinetic Monte Carlo Molecular Dynamics) emprunte une voie différente. Il laisse les simulations conventionnelles gérer le mouvement moléculaire, puis utilise un module supplémentaire pour chercher les lieux où des réactions pourraient se produire, estimer leur vitesse et choisir aléatoirement quelle réaction se produit ensuite. En répétant les cycles mouvement — recherche de réaction — choix de réaction, KIMMDY peut suivre des cascades réactionnelles complexes sur des durées allant de fractions de seconde à beaucoup plus long, sans jamais calculer un chemin réactionnel complet au niveau quantique.
Comment fonctionne l'émulateur de réactions
Au cœur de KIMMDY se trouve un « émulateur de réactions » qui prédit des vitesses réactionnelles à partir des formes instantanées des molécules. Pour chaque instantané issu d'une simulation de dynamique moléculaire, KIMMDY identifie les partenaires potentiels et utilise différents modèles pour estimer l'amplitude de la barrière pour chaque réaction. Dans de nombreux cas, il recourt à des réseaux de neurones graphiques — des modèles d'apprentissage automatique qui traitent les atomes comme des nœuds et les liaisons comme des liens — pour deviner les hauteurs de barrières à partir de calculs quantiques de haut niveau effectués auparavant. Dans d'autres cas, il se rabat sur des formules physiques plus simples ou des règles heuristiques tirées d'expériences. À partir de ces barrières, il calcule des taux et les injecte dans une étape de Monte Carlo cinétique qui choisit aléatoirement la réaction suivante, avance l'horloge simulée, puis met à jour la structure moléculaire et le champ de forces avant le prochain cycle de mouvement moléculaire.
Suivre les dommages radicaux dans les échafaudages protéiques
Pour vérifier que KIMMDY fonctionne comme prévu, les auteurs l'ont d'abord appliqué à une classe simple de réactions dans de petits radicaux organiques pour lesquels des données expérimentales existent. L'émulateur a reproduit quelles réactions de transfert d'hydrogène sont les plus probables, même s'il a sous-estimé leurs vitesses absolues. Ils sont ensuite passés à un cas beaucoup plus complexe : le collagène, la protéine fibreuse qui confère force aux tissus conjonctifs. Sous contrainte mécanique, des liaisons dans le collagène peuvent se rompre de manière homolytique, créant des radicaux très réactifs qui sautent d'un site à l'autre en transférant des atomes d'hydrogène le long d'une chaîne. Dans un fibrille de collagène composé de 2,6 millions d'atomes, KIMMDY a tracé des centaines de ces sauts et montré que les radicaux peuvent migrer à plusieurs nanomètres de leur origine. Les simulations ont révélé qu'une réticulation inhabituelle (appelée PYD) et un acide aminé modifié (DOPA) agissent tous deux comme des pièges de radicaux exceptionnellement efficaces, une conclusion étayée par des calculs thermodynamiques et par la réinterprétation de spectres antérieurs de résonance paramagnétique électronique.
Quand différentes réactions entrent en compétition
De nombreux systèmes biologiques peuvent rompre la même liaison de plusieurs manières, conduisant soit à la formation de radicaux, soit à des produits « à coque fermée » plus conventionnels. Dans le collagène, par exemple, une liaison peptidique peut soit se scinder symétriquement en deux radicaux (homolyse), soit réagir avec l'eau et se fendre de façon inégale (hydrolyse). Les simulations quantiques directes des deux options dans un environnement réaliste seraient prohibitives. Avec KIMMDY, les auteurs ont combiné un modèle physique pour l'homolyse assistée par la force avec un modèle heuristique, fondé sur l'expérience, pour l'hydrolyse, et ont laissé les deux réactions entrer en compétition à la fois dans un peptide isolé et dans un fibrille de collagène dense. Ils ont constaté que dans une chaîne isolée soumise à traction, l'hydrolyse domine clairement. Dans un fibrille encombré et réticulé, cependant, la contrainte mécanique se concentre si fortement dans certaines régions que l'homolyse devient compétitive voire plus rapide, expliquant pourquoi la formation de radicaux est observée dans les tissus réels.
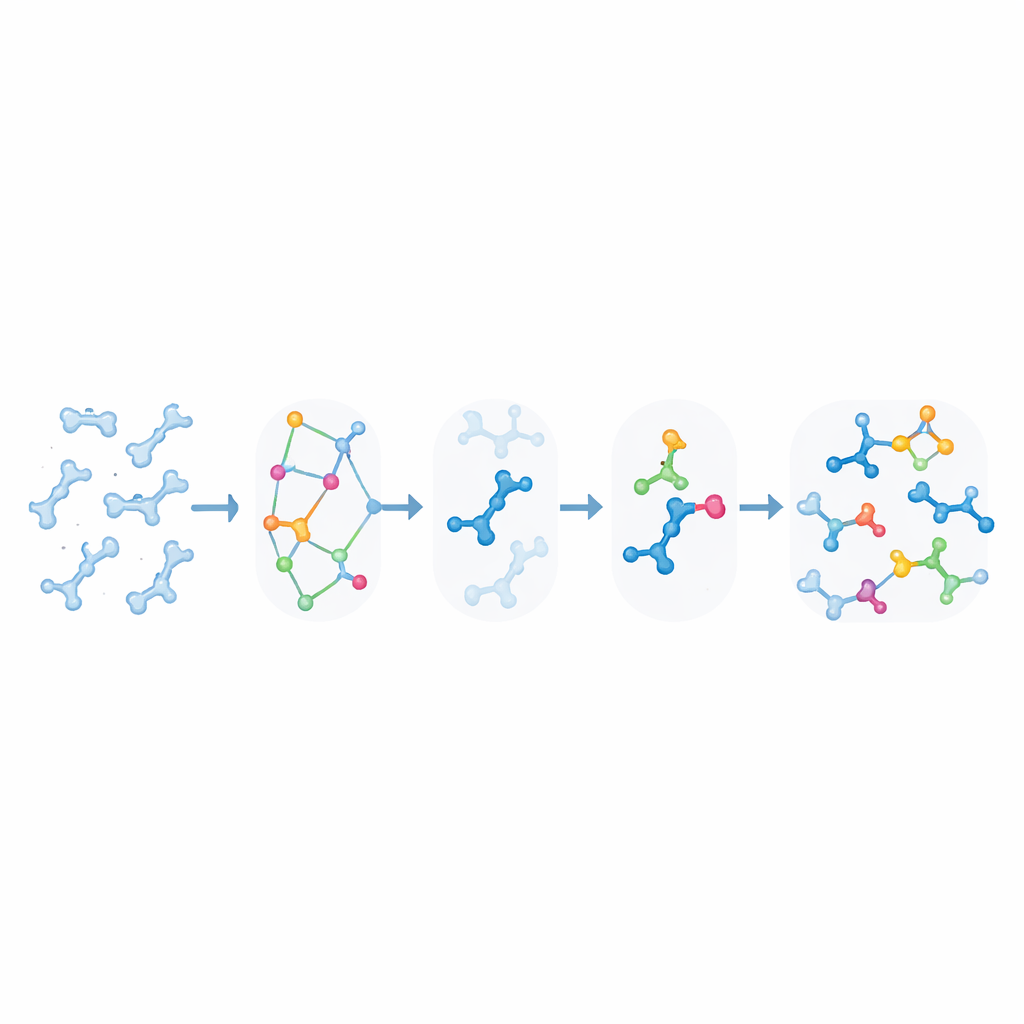
Dommages induits par la lumière et réticulations dans l'ADN
KIMMDY éclaire également la manière dont les ultraviolets modifient l'ADN. Lorsque deux thymines voisines absorbent des UV, elles peuvent fusionner en un dimère cyclobutane, une lésion fortement liée au cancer de la peau mais aussi exploitée pour réticuler des brins en nanotechnologie de l'ADN. En utilisant une règle géométrique simple qui relie distance et angle entre liaisons réactives à la probabilité de réaction, ajustée pour correspondre à des expériences sur de petites molécules, les auteurs ont employé KIMMDY pour estimer des « rendements quantiques » — la fréquence de formation d'un dimère par photon absorbé — pour divers motifs d'ADN. L'émulateur a prédit que certains agencements couramment utilisés en origami d'ADN, comme des surplombs et des croisements, présentent en réalité des rendements surprisamment faibles comparés à l'ADN double brin standard ou à l'ADN nické. Il a aussi suggéré que la formation d'un dimère n'altère pas de manière significative les chances d'en former un autre à proximité, impliquant que des dommages antérieurs ne prédisposent pas nécessairement l'ADN à d'autres dimères.
Pourquoi cela compte pour la biologie et la conception
En termes simples, KIMMDY offre une façon rapide et flexible d'explorer des scénarios « et si » pour la chimie au sein de la matière vivante. En émulant plutôt qu'en simulant pleinement les réactions, il peut gérer des systèmes énormes et des échelles de temps longues tout en tenant compte de la manière dont les formes sans cesse changeantes des molécules influent sur quelles réactions surviennent, où et quand. La méthode a aidé à découvrir un site de stabilisation des radicaux jusque-là négligé dans le collagène, a clarifié comment les forces mécaniques déplacent l'équilibre entre voies de rupture de liaison, et a soulevé de nouvelles questions sur la formation et la localisation des réticulations d'ADN sous l'effet de la lumière. À mesure que l'émulateur s'étend à plus de types de réactions et à des modèles de taux plus précis, il promet de devenir un outil puissant pour comprendre comment les biomolécules vieillissent, dysfonctionnent ou peuvent être conçues pour la médecine et la nanotechnologie.
Citation: Hartmann, E., Buhr, J., Riedmiller, K. et al. KIMMDY: a biomolecular reaction emulator. Nat Commun 17, 3500 (2026). https://doi.org/10.1038/s41467-026-71955-2
Mots-clés: simulations biomoléculaires, cinétique des réactions, modélisation moléculaire, dommages de l'ADN