Clear Sky Science · sv
Ursprung och omfång av parvis epistas i en α-helix i β-laktamasen TEM-1
Varför små förändringar i ett protein spelar roll för antibiotikaresistens
Antibiotikaresistenta bakterier utgör ett växande hot, och många av dem förlitar sig på enzymer som bryter ner läkemedel innan de hinner verka. Ett sådant enzym, kallat TEM-1 β-laktamas, hjälper vanliga tarmbakterier att stå emot ofta använda antibiotika som amoxicillin. Denna studie zoomar in på en mycket liten del av det enzymet — en 11–aminosyraspiral kallad en α-helix — för att ställa en stor fråga: hur kombineras par av mutationer för att göra resistens lättare eller svårare att utveckla, och kan vi förutsäga dessa effekter utifrån grundläggande fysikaliska principer och evolutionära data?
När en mutation beror på en annan
Huvudidén som undersöks här är ”epistasis”, vilket betyder att effekten av en mutation beror på vilka andra mutationer som finns närvarande. Istället för att verka oberoende kan mutationer hjälpa eller stjälpa varandra på överraskande sätt. Författarna byggde ett gigantiskt bibliotek med mer än 14 000 varianter av TEM-1-enzymet, där varje variant bar antingen en eller två förändringar inom en enda α-helix som inte är en del av det aktiva sätet men som hjälper till att behålla proteinets form. De mätte sedan hur väl varje variant skyddade bakterier mot amoxicillin på två sätt: genom att följa bakteriernas tillväxt i konkurrens (en direkt mått på fitness) och genom att bestämma den minsta antibiotikakoncentration som förhindrar tillväxt (MIC, ett standardkliniskt mått på resistens). Dessa parade mätningar gjorde det möjligt att kvantifiera hur ofta mutationseffekter är kontextberoende snarare än helt additiva.
Många mutationer förstör enzymet, men de flesta interaktioner är förutsägbara
Det första slående resultatet är hur känslig denna lilla helix är. Nästan hälften av alla singelmutationer i området förstörde i praktiken enzymets funktion under testförhållandena, och nästan fyra av fem dubbelmutanter var icke-funktionella. Vissa utbyten, som införandet av aminosyran prolin, var nästan alltid dödligt eftersom de stör helixens form. När teamet undersökte hur dubbelmutanter uppträdde jämfört med vad som skulle förväntas från att helt enkelt lägga ihop effekterna av varje singelmutation fann de utbredd epistasis: när riktigt icke-funktionella varianter uteslöts visade ungefär 90 % av mutationerna tydligt kontextberoende. Dock hade det mesta av denna epistasis ett enkelt mönster — mutationer tenderade att se värre ut på redan försvagade bakgrunder, en form av ”global” negativ interaktion.
En enkel veckningsmodell förklarar det mesta av komplexiteten
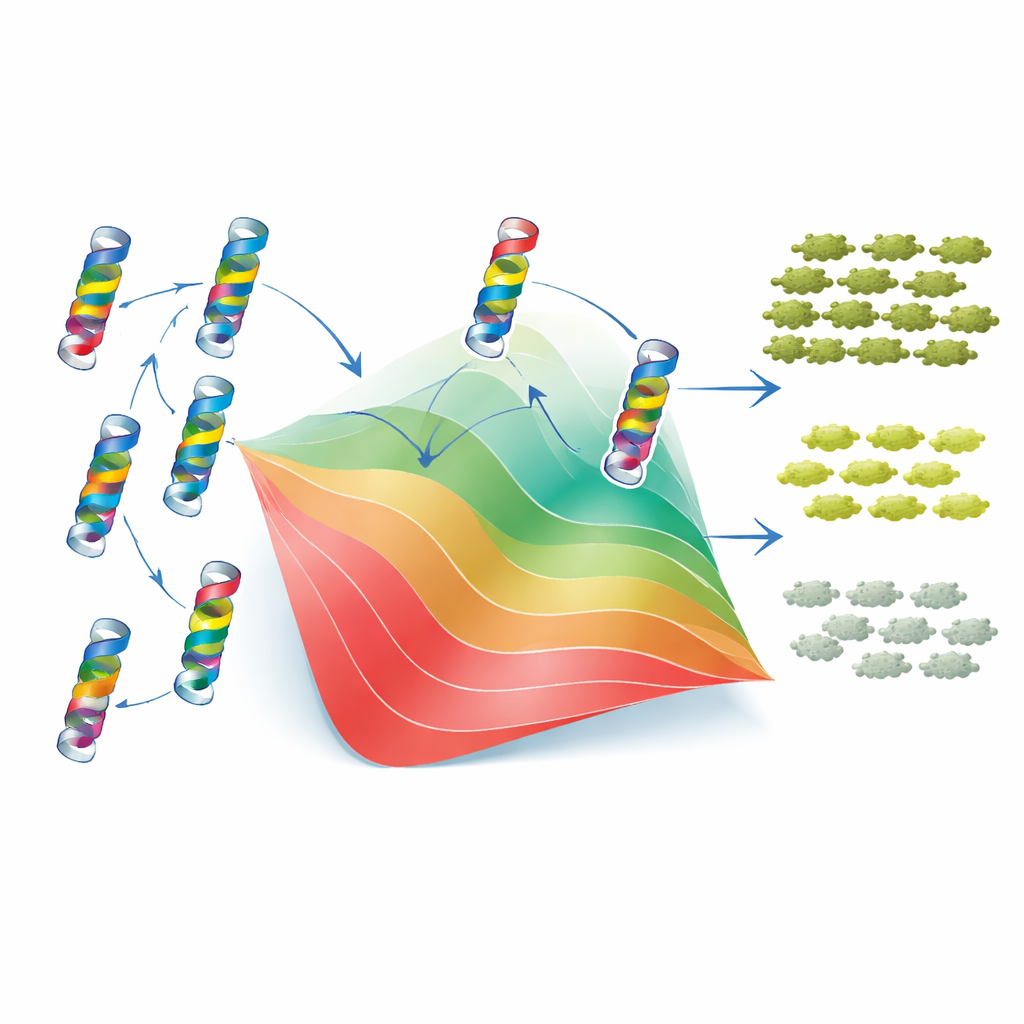
För att förstå varför dessa mönster uppstår vände sig författarna till en klassisk tvåtillståndsmodell av proteinveckning. I denna syn är TEM-1 antingen vikt och funktionell eller ovikt och värdelös, och varje mutation skjuter balansen mellan dessa tillstånd genom att höja eller sänka proteinets stabilitet. Avgörande är att modellen antar att stabilitetsförändringarna från enskilda mutationer adderas, även om den slutliga effekten på fitness är icke-linjär: när proteinet väl är tillräckligt instabil får små ytterligare förändringar mycket större konsekvenser. Genom att anpassa detta ramverk till sina data kunde forskarna tilldela varje mutation en effektiv ”stabilitetskostnad” eller ”stabilitetsvinst”. Med bara dessa värden och en enda baslinjestabilitetsparameter reproducerade modellen nära nog både den uppmätta fitnessen och MIC för tusentals singel- och dubbelmutanter, liksom den övergripande spridningen och snedheten i de epistatiska interaktionerna. Med andra ord uppstår mycket av den uppenbara komplexiteten i mutationsinteraktioner från en enkel fysisk begränsning: behovet av att proteinet förblir vikt.
Lokala kontakter och långsiktig evolution lämnar ett finare avtryck
Berättelsen är dock inte helt global. Tvåtillståndsmodellen fungerade sämre för par av restpositioner som sitter mycket nära varandra i enzymets tredimensionella struktur. För sådana grannar avslöjade data ”idiosynkratisk” epistasis — specifika kombinationer som uppträdde bättre eller sämre än förväntat från stabilitet ensam, ibland genom att kompensera varandras laddning eller form. För att se om evolutionen bevarat spår av dessa lokala interaktioner analyserade författarna tusentals relaterade β-laktamassekvenser med en statistisk ”Potts”-modell som fångar vilka rester som tenderar att variera tillsammans över arter. Dessa sekvensbaserade kopplingar stämde på två sätt överens med de experimentella fynden: de korrelerade med de härledda stabilitetseffekterna och framhävde många av samma restpar där den enkla veckningsmodellen misslyckades. När Potts-modellens utdata noggrant omskalades och fördes genom samma veckningsramverk kunde de till och med delvis förutsäga tecknet på epistasis för nya mutationspar.
Vad detta betyder för att förutsäga resistens
För icke-specialister är huvudbudskapet att även i ett litet strukturellt element av ett enda protein är interaktioner mellan mutationer vanliga men inte slumpmässiga. De flesta kan förstås som följder av hur mutationer gemensamt skjuter enzymet mot eller bort från ett stabilt vikt tillstånd, medan en minoritet speglar direkta lokala kontakter som finjusterar beteendet. Det faktum att båda typerna av effekter lämnar upptäckbara spår i β-laktamasernas långsiktiga evolution tyder på att reglerna för mutationsinteraktioner är tillräckligt stabila för att läras in och generaliseras. Detta väcker möjligheten att, genom att kombinera djupa muteringsskanningar med modeller för proteinveckning och evolutionära sekvensdata, vi så småningom kan förutsäga hur resistensenzym kommer att förändras — och utforma läkemedel eller terapier som är svårare för dem att överlista.
Citering: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Nyckelord: epistasis, proteinstabilitet, beta-laktamas, antibiotikaresistens, djup muteringsskanning