Clear Sky Science · it
Origine e portata dell'epistasi a coppie in un α-elica della β-lattamasi TEM-1
Perché piccole modifiche in una proteina contano per la resistenza agli antibiotici
I batteri resistenti agli antibiotici rappresentano una minaccia crescente e molti di essi si affidano ad enzimi che inattivano i farmaci prima che possano agire. Un enzima di questo tipo, chiamato β-lattamasi TEM-1, aiuta batteri intestinali comuni a resistere ad antibiotici ampiamente usati come l’amoxicillina. Questo studio si concentra su una parte molto piccola dell’enzima — un’elica α di 11 amminoacidi — per porre una domanda ambiziosa: come si combinano coppie di mutazioni per rendere la resistenza più o meno facile da evolvere, e possiamo prevedere questi effetti a partire da principi fisici di base e da dati evolutivi?
Quando una mutazione dipende da un’altra
L’idea centrale esplorata è l’“epistasi”, che significa che l’effetto di una mutazione dipende da quali altre mutazioni sono presenti. Invece di agire in modo indipendente, le mutazioni possono aiutarsi o ostacolarsi a vicenda in modi inaspettati. Gli autori hanno costruito una libreria enorme di oltre 14.000 varianti dell’enzima TEM-1, ciascuna contenente una o due modifiche all’interno della stessa α-elica che non fa parte del sito attivo ma contribuisce a mantenere la struttura della proteina. Hanno quindi misurato quanto ciascuna variante proteggeva i batteri dall’amoxicillina in due modi: monitorando la crescita batterica in competizione (una misura diretta della fitness) e determinando la concentrazione minima di antibiotico che impedisce la crescita (la MIC, una misura clinica standard della resistenza). Queste misure abbinate hanno permesso di quantificare quanto spesso gli effetti delle mutazioni dipendono dal contesto invece di sommarsi semplicemente.
Molte mutazioni compromettono l’enzima, ma la maggior parte delle interazioni è prevedibile
Il primo risultato sorprendente è la fragilità di questa piccola elica. Quasi la metà di tutte le mutazioni singole nella regione hanno di fatto distrutto la funzione dell’enzima nelle condizioni di prova, e quasi quattro mutanti doppi su cinque erano non funzionali. Alcune sostituzioni, come l’introduzione della prolina, erano quasi sempre letali perché perturbano la forma dell’elica. Quando il team ha confrontato il comportamento dei doppi mutanti con quanto ci si sarebbe aspettati sommando gli effetti di ciascuna mutazione singola, ha riscontrato un’epistasi diffusa: escluse le varianti effettivamente inattive, circa il 90% delle mutazioni mostrava una chiara dipendenza dal contesto. Tuttavia, la maggior parte di questa epistasi seguiva un modello semplice — le mutazioni tendevano a risultare più dannose su sfondi già indeboliti, una forma di interazione negativa “globale”.
Un semplice modello di folding spiega gran parte della complessità
Per capire perché emergono questi schemi, gli autori hanno utilizzato il classico modello a due stati del folding proteico. In questa visione, TEM-1 è o ripiegata e funzionale o denaturata e inutile, e ogni mutazione sposta l’equilibrio tra questi stati aumentando o diminuendo la stabilità della proteina. Cruciale è l’assunto che le variazioni di stabilità dovute a mutazioni individuali si sommano, anche se l’effetto finale sulla fitness è non lineare: una volta che la proteina è sufficientemente instabile, piccoli ulteriori cambiamenti hanno conseguenze molto più grandi. Adattando questo quadro ai loro dati, i ricercatori hanno potuto assegnare a ogni mutazione un costo o un guadagno efficace di “stabilità”. Con solo questi valori e un singolo parametro di stabilità di base, il modello ha riprodotto da vicino sia la fitness misurata sia la MIC di migliaia di mutanti singoli e doppi, così come la distribuzione e l’asimmetria complessiva delle interazioni epistatiche. In altre parole, buona parte della complessità apparente delle interazioni tra mutazioni deriva da un vincolo fisico semplice: la necessità che la proteina rimanga ripiegata.
Contatti locali e l’evoluzione a lungo termine lasciano un’impronta più sottile
La storia non è però puramente globale. Il modello a due stati funzionava meno bene per coppie di residui che si trovano molto vicini nella struttura 3D dell’enzima. Per questi vicini, i dati hanno rivelato un’epistasi “idiosincratica” — combinazioni specifiche che si comportavano meglio o peggio di quanto previsto dalla sola stabilità, a volte compensandosi a vicenda per carica o forma. Per verificare se l’evoluzione aveva preservato tracce di queste interazioni locali, gli autori hanno analizzato migliaia di sequenze correlate di β-lattamasi usando un modello statistico “Potts” che cattura quali residui tendono a variare insieme tra le specie. Questi accoppiamenti basati sulle sequenze si sono allineati in due modi con i risultati sperimentali: hanno correlato con gli effetti di stabilità dedotti e hanno evidenziato molte delle stesse coppie di residui dove il semplice modello di folding falliva. Quando le uscite del modello Potts sono state opportunamente ridimensionate e immesse nello stesso quadro di folding, hanno persino potuto prevedere in parte il segno dell’epistasi per nuove coppie di mutazioni.
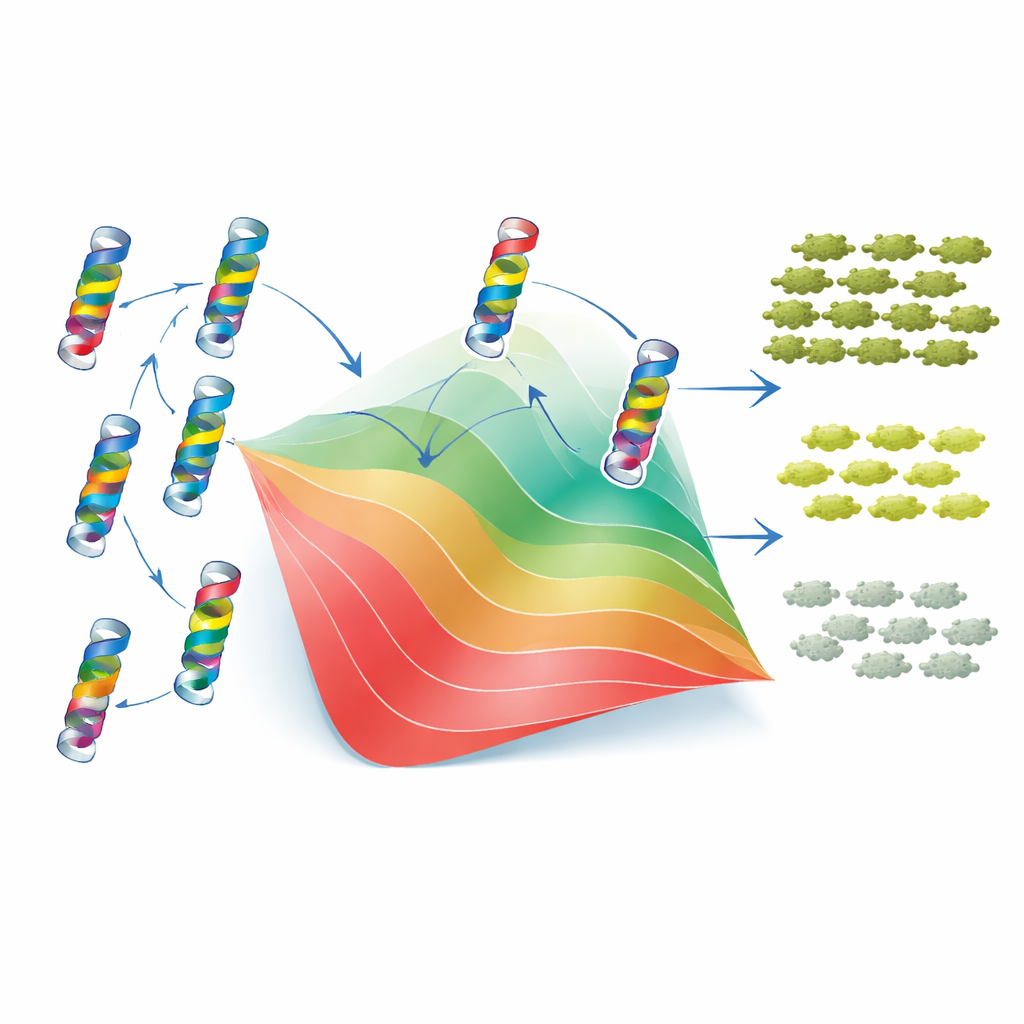
Cosa significa per prevedere la resistenza
Per i non specialisti, la conclusione principale è che anche in un elemento strutturale minuscolo di una singola proteina, le interazioni tra mutazioni sono comuni ma non casuali. La maggior parte può essere interpretata come conseguenza del modo in cui le mutazioni spingono collettivamente l’enzima verso o lontano da uno stato ripiegato stabile, mentre una minoranza riflette contatti locali diretti che rifiniscono il comportamento. Il fatto che entrambi i tipi di effetti lascino tracce rilevabili nell’evoluzione a lungo termine delle β-lattamasi suggerisce che le regole delle interazioni mutazionali sono sufficientemente stabili da poter essere apprese e generalizzate. Ciò apre la possibilità che, combinando deep mutational scan con modelli di folding proteico e dati di sequenze evolutive, si possa infine prevedere come cambieranno gli enzimi della resistenza — e progettare farmaci o terapie più difficili da eludere per loro.
Citazione: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Parole chiave: epistasi, stabilità proteica, beta-lattamasi, resistenza agli antibiotici, deep mutational scanning