Clear Sky Science · pl
Pochodzenie i zakres epistazy parowej w α-helisie β-laktamazy TEM-1
Dlaczego drobne zmiany w białku mają znaczenie dla oporności na antybiotyki
Bakterie odporne na antybiotyki stanowią rosnące zagrożenie, a wiele z nich polega na enzymach, które rozkładają leki, zanim te zdążą zadziałać. Jednym z takich enzymów jest TEM-1 β-laktamaza, która pomaga powszechnym bakteriom jelitowym przetrwać powszechnie stosowane antybiotyki, takie jak amoksycylina. Niniejsze badanie przygląda się bardzo małej części tego enzymu — 11-aminokwasowej spirali zwanej α-helisą — aby postawić szerokie pytanie: jak pary mutacji łączą się, by ułatwiać lub utrudniać ewolucję oporności i czy można przewidzieć te efekty na podstawie podstawowych zasad fizycznych oraz danych ewolucyjnych?
Kiedy jedna mutacja zależy od innej
Główny koncept badany tutaj to „epistaza”, co oznacza, że efekt jednej mutacji zależy od obecności innych mutacji. Zamiast działać niezależnie, mutacje mogą wzajemnie sobie pomagać lub przeszkadzać w zaskakujący sposób. Autorzy zbudowali ogromną bibliotekę ponad 14 000 wariantów enzymu TEM-1, z których każdy nosił jedną lub dwie zmiany w obrębie pojedynczej α-helisy, która nie wchodzi w skład miejsca aktywnego, ale pomaga utrzymać kształt białka. Następnie zmierzyli, jak dobrze każdy wariant chronił bakterie przed amoksycyliną, na dwa sposoby: śledząc wzrost bakterii w konkurencji (bezpośredni miernik fitnessu) oraz określając minimalne stężenie antybiotyku, które hamuje wzrost (MIC, standardowy kliniczny wskaźnik oporności). Te parowane pomiary pozwoliły oszacować, jak często efekty mutacji są zależne od kontekstu, zamiast być prostą sumą efektów pojedynczych mutacji.
Wiele mutacji niszczy enzym, ale większość interakcji jest przewidywalna
Pierwszy uderzający wynik pokazuje, jak krucha jest ta mała helisa. Prawie połowa wszystkich pojedynczych mutacji w tym regionie praktycznie niszczyła funkcję enzymu w warunkach testowych, a niemal cztery na pięć mutantów z dwiema mutacjami były nieaktywne. Niektóre podstawienia, takie jak wprowadzenie proliny, były niemal zawsze śmiertelne, ponieważ zaburzają kształt helisy. Gdy zespół porównał zachowanie mutantów podwójnych z oczekiwaniami wynikającymi z prostego dodawania efektów pojedynczych mutacji, ujawniła się szeroka epistaza: po wyłączeniu naprawdę nieaktywnych wariantów około 90% mutacji wykazywało wyraźną zależność od tła. Jednak większość tej epistazy miała prosty wzorzec — mutacje zwykle wyglądały gorzej na już osłabionym tle, forma „globalnej” negatywnej interakcji.
Prosty model fałdowania wyjaśnia większość złożoności
Aby zrozumieć, dlaczego pojawiają się te wzorce, autorzy odwołali się do klasycznego dwustanowego modelu fałdowania białka. W tym ujęciu TEM-1 jest albo złożony i funkcjonalny, albo rozłożony i bezużyteczny, a każda mutacja przesuwa równowagę między tymi stanami poprzez podnoszenie lub obniżanie stabilności białka. Kluczowe jest założenie, że zmiany stabilności wywołane przez pojedyncze mutacje sumują się, mimo że ostateczny wpływ na fitness jest nieliniowy: gdy białko staje się wystarczająco niestabilne, niewielkie dodatkowe zmiany mają znacznie większe konsekwencje. Dopasowując ten model do swoich danych, badacze mogli przypisać każdej mutacji efektywny „koszt stabilności” lub „zysk stabilności”. Mając tylko te wartości i jeden parametryczny poziom stabilności wyjściowej, model wiernie odtworzył zarówno zmierzony fitness, jak i MIC dla tysięcy pojedynczych i podwójnych mutantów, a także ogólny rozkład i tendencję interakcji epistatycznych. Innymi słowy, wiele pozornej złożoności interakcji mutacji wynika z prostego ograniczenia fizycznego: konieczności utrzymania białka w stanie złożonym.
Lokalne kontakty i długoterminowa ewolucja zostawiają subtelniejszy ślad
Opowieść nie jest jednak wyłącznie globalna. Dwustanowy model radził sobie gorzej z parami reszt, które leżą bardzo blisko siebie w strukturze 3D enzymu. Dla takich sąsiadów dane ujawniły „idiozyncraficzną” epistazę — specyficzne kombinacje zachowywały się lepiej lub gorzej niż wynikałoby to z samej stabilności, czasem kompensując wzajemnie ładunek lub kształt. Aby sprawdzić, czy ewolucja zachowała ślady tych lokalnych interakcji, autorzy przeanalizowali tysiące powiązanych sekwencji β-laktamaz przy użyciu statystycznego modelu „Pottsa”, który wychwytuje, które reszty mają tendencję do współzmienności między gatunkami. Te oparte na sekwencjach sprzężenia zgadzały się z wynikami eksperymentalnymi na dwa sposoby: korelowały z wnioskowanymi efektami stabilności i wskazywały wiele tych samych par reszt, dla których prosty model fałdowania zawodził. Gdy wyjścia modelu Pottsa zostały odpowiednio przeskalowane i przepuszczone przez ten sam ramowy model fałdowania, mogły nawet częściowo przewidzieć znak epistazy dla nowych par mutacji.
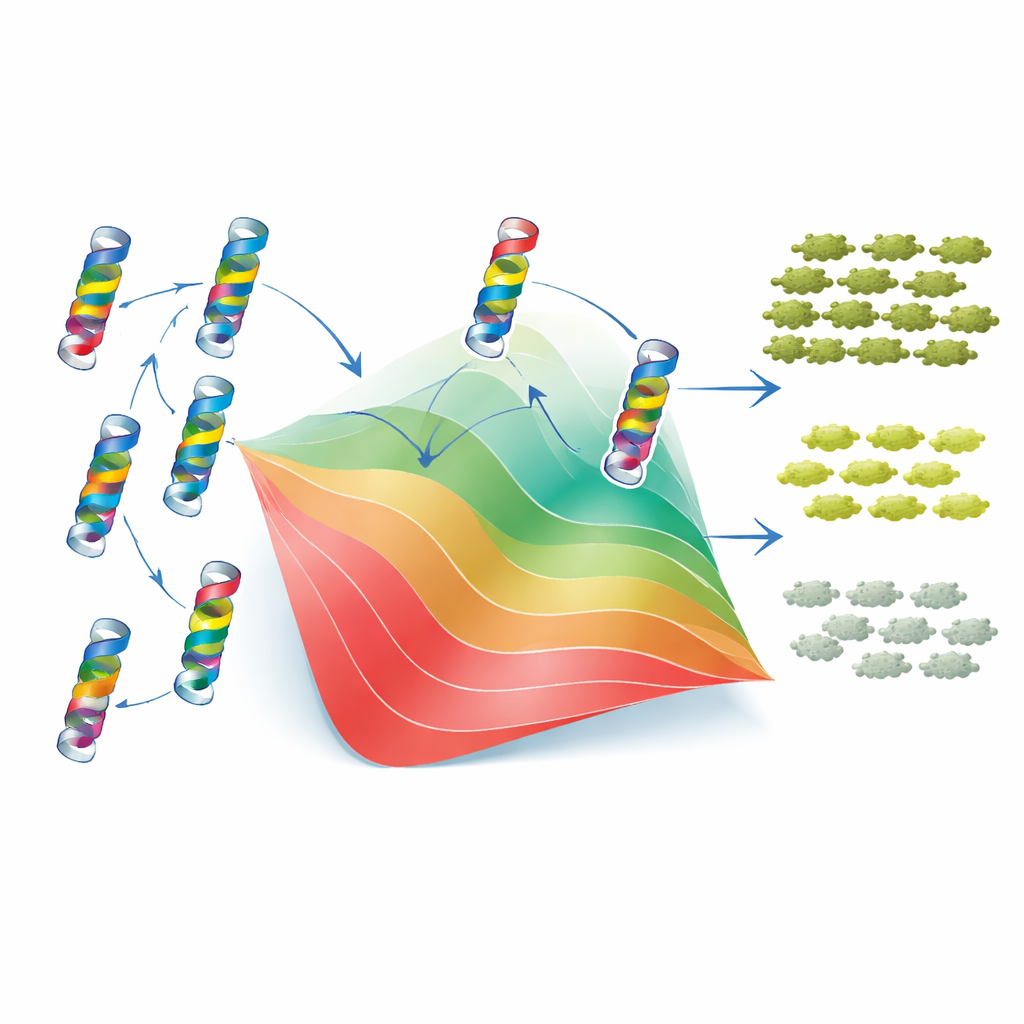
Co to oznacza dla przewidywania oporności
Dla czytelników niebędących specjalistami najważniejsze wnioski są takie, że nawet w maleńkim elemencie strukturalnym pojedynczego białka interakcje między mutacjami są powszechne, ale nie losowe. Większość można zrozumieć jako konsekwencję tego, jak mutacje łącznie przesuwają enzym w stronę lub z dala od stabilnego stanu złożonego, podczas gdy mniejszość odzwierciedla bezpośrednie lokalne kontakty, które dopracowują zachowanie. Fakt, że oba typy efektów zostawiają wykrywalne ślady w długoterminowej ewolucji β-laktamaz sugeruje, że zasady interakcji mutacyjnych są na tyle stabilne, że można je poznać i uogólnić. To otwiera możliwość, że łącząc głębokie skany mutacyjne z modelami fałdowania białek i danymi sekwencji ewolucyjnej, w przyszłości będziemy w stanie przewidzieć, jak enzymy oporności będą się zmieniać — i projektować leki lub terapie, którym trudniej będzie się przeciwstawić.
Cytowanie: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Słowa kluczowe: epistaza, stabilność białka, beta-laktamaza, oporność na antybiotyki, głębokie skanowanie mutacyjne