Clear Sky Science · fr
Origines et étendue de l’épistasie par paires dans une α-hélice de la β-lactamase TEM-1
Pourquoi de minuscules changements dans une protéine comptent pour la résistance aux antibiotiques
Les bactéries résistantes aux antibiotiques représentent une menace croissante, et bon nombre d’entre elles dépendent d’enzymes qui dégradent les médicaments avant qu’ils n’agissent. Une de ces enzymes, appelée β-lactamase TEM-1, aide des bactéries intestinales courantes à résister à des antibiotiques largement utilisés comme l’amoxicilline. Cette étude se concentre sur une toute petite partie de cette enzyme — une spirale de 11 acides aminés appelée α-hélice — pour poser une grande question : comment des paires de mutations se combinent-elles pour faciliter ou compliquer l’évolution de la résistance, et peut-on prédire ces effets à partir de principes physiques de base et de données évolutives ?
Quand une mutation dépend d’une autre
L’idée centrale explorée ici est l’« épistasie », qui signifie que l’effet d’une mutation dépend des autres mutations présentes. Plutôt que d’agir de manière indépendante, les mutations peuvent se renforcer ou s’affaiblir mutuellement de façon surprenante. Les auteurs ont construit une vaste bibliothèque de plus de 14 000 variantes de l’enzyme TEM-1, chacune portant soit une, soit deux modifications au sein d’une même α-hélice qui ne fait pas partie du site actif mais contribue à maintenir la conformation de la protéine. Ils ont ensuite mesuré la capacité de chaque variante à protéger les bactéries contre l’amoxicilline de deux façons : en suivant la croissance concurrentielle des bactéries (une mesure directe de la fitness) et en déterminant la concentration minimale d’antibiotique empêchant la croissance (la CMI, une mesure clinique standard de la résistance). Ces mesures jumelées leur ont permis de quantifier à quelle fréquence les effets des mutations dépendent du contexte plutôt que d’être simplement additifs.
Beaucoup de mutations détruisent l’enzyme, mais la plupart des interactions sont prévisibles
Le premier résultat frappant est la fragilité de cette petite hélice. Près de la moitié de toutes les mutations simples dans la région ont essentiellement détruit la fonction de l’enzyme dans les conditions testées, et presque quatre mutants doubles sur cinq étaient non fonctionnels. Certaines substitutions, comme l’introduction de la proline, étaient presque toujours létales car elles perturbent la conformation de l’hélice. Lorsque l’équipe a comparé le comportement des mutants doubles à ce qui serait attendu en additionnant simplement les effets de chaque mutation simple, elle a observé une épistasie répandue : une fois que les variantes véritablement inactives ont été exclues, environ 90 % des mutations montraient une dépendance claire au contexte. Cependant, la majeure partie de cette épistasie suivait un schéma simple — les mutations avaient tendance à paraître plus délétères sur des fonds déjà affaiblis, une forme d’interaction « globale » négative.
Un modèle simple de repliement explique la majeure partie de la complexité
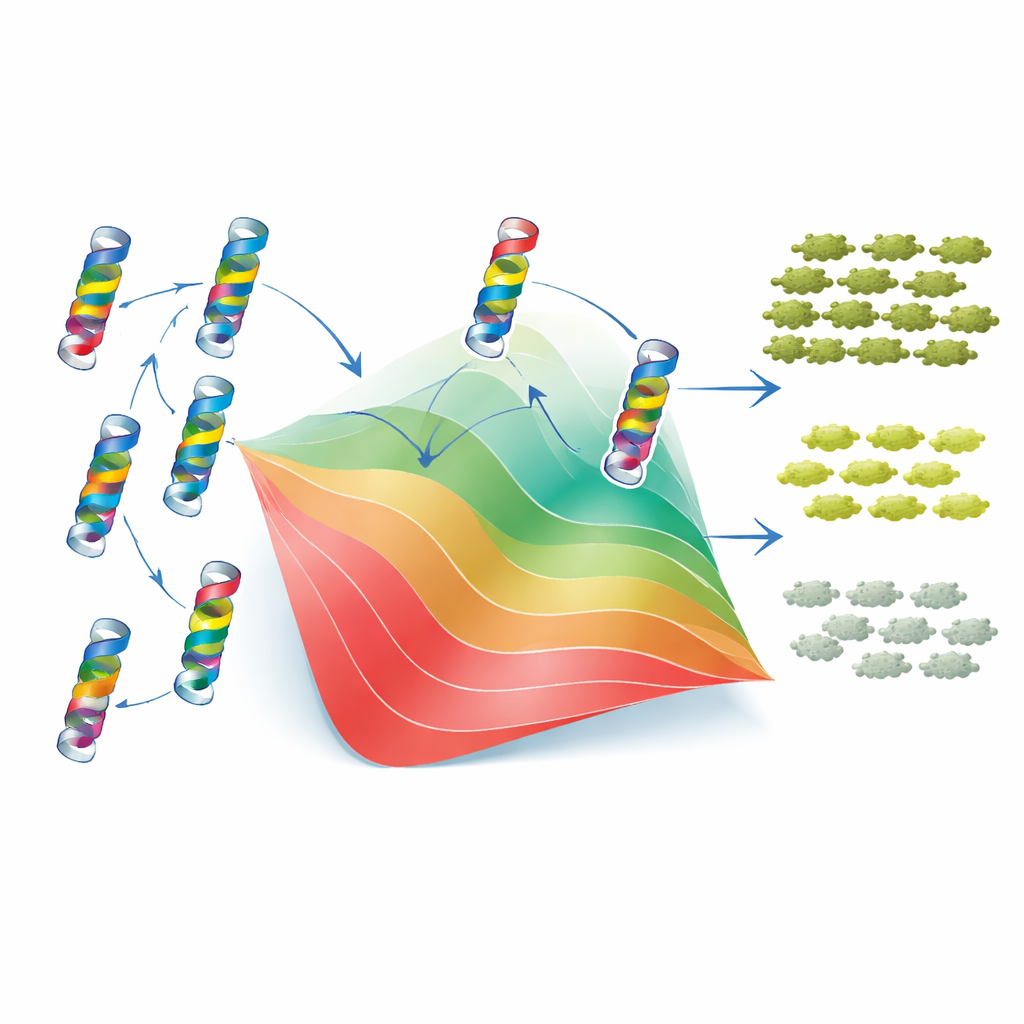
Pour comprendre pourquoi ces motifs apparaissent, les auteurs ont recouru à un modèle classique à deux états du repliement des protéines. Dans cette perspective, TEM-1 est soit repliée et fonctionnelle, soit dépliée et inutile, et chaque mutation déplace l’équilibre entre ces états en abaissant ou en augmentant la stabilité de la protéine. De façon cruciale, le modèle suppose que les changements de stabilité dus aux mutations individuelles s’additionnent, même si l’effet final sur la fitness est non linéaire : une fois que la protéine est suffisamment instable, de petits changements supplémentaires ont des conséquences beaucoup plus importantes. En ajustant ce cadre sur leurs données, les chercheurs ont pu attribuer à chaque mutation un « coût » ou un « gain » effectif de stabilité. Avec seulement ces valeurs et un seul paramètre de stabilité de référence, le modèle a reproduit de près à la fois la fitness mesurée et la CMI de milliers de mutants simples et doubles, ainsi que la distribution et l’asymétrie globales des interactions épistatiques. Autrement dit, une grande partie de la complexité apparente des interactions entre mutations découle d’une contrainte physique simple : la nécessité pour la protéine de rester repliée.
Les contacts locaux et l’évolution à long terme laissent une empreinte plus subtile
L’histoire n’est toutefois pas entièrement globale. Le modèle à deux états fonctionnait moins bien pour les paires de résidus très proches dans la structure 3D de l’enzyme. Pour ces voisins, les données révélaient une épistasie « idiosyncratique » — des combinaisons spécifiques qui se comportaient mieux ou pire que prévu par la stabilité seule, compensant parfois mutuellement les charges ou la géométrie. Pour savoir si l’évolution avait préservé des signatures de ces interactions locales, les auteurs ont analysé des milliers de séquences de β-lactamases apparentées en utilisant un modèle statistique de type « Potts » qui capture quels résidus tendent à varier ensemble entre les espèces. Ces couplages fondés sur les séquences s’alignaient avec les résultats expérimentaux de deux manières : ils corrélaient avec les effets de stabilité déduits et mettaient en évidence nombre des mêmes paires de résidus où le modèle simple de repliement échouait. Lorsque les sorties du modèle Potts étaient soigneusement re‑mise à l’échelle et intégrées dans le même cadre de repliement, elles pouvaient même en partie prédire le signe de l’épistasie pour de nouvelles paires de mutations.
Ce que cela signifie pour prédire la résistance
Pour les non‑spécialistes, l’enseignement principal est que, même dans un tout petit élément structurel d’une seule protéine, les interactions entre mutations sont fréquentes mais pas aléatoires. La plupart peuvent être comprises comme les conséquences de la façon dont les mutations poussent collectivement l’enzyme vers un état replié plus ou moins stable, tandis qu’une minorité reflète des contacts locaux directs qui affinent le comportement. Le fait que les deux types d’effets laissent des traces détectables dans l’évolution à long terme des β-lactamases suggère que les règles d’interaction mutuelle sont suffisamment stables pour être apprises et généralisées. Cela ouvre la possibilité que, en combinant des balayages mutagènes profonds avec des modèles de repliement protéique et des données de séquences évolutives, nous puissions un jour prédire comment les enzymes de résistance évolueront — et concevoir des médicaments ou des thérapies plus difficiles à contourner pour elles.
Citation: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Mots-clés: épistasie, stabilité des protéines, β-lactamase, résistance aux antibiotiques, balayage mutagène profond