Clear Sky Science · es
Orígenes y alcance de la epistasis por pares en una α-hélice de la β-lactamasa TEM-1
Por qué cambios diminutos en una proteína importan para la resistencia a antibióticos
Las bacterias resistentes a antibióticos son una amenaza creciente, y muchas dependen de enzimas que inactivan los fármacos antes de que puedan actuar. Una de estas enzimas, llamada β-lactamasa TEM-1, ayuda a bacterias intestinales comunes a resistir antibióticos ampliamente usados como la amoxicilina. Este estudio se centra en una parte muy pequeña de esa enzima —una espiral de 11 aminoácidos llamada α-hélice— para plantear una gran pregunta: ¿cómo se combinan pares de mutaciones para facilitar o dificultar la evolución de la resistencia, y podemos predecir estos efectos a partir de principios físicos básicos y de datos evolutivos?
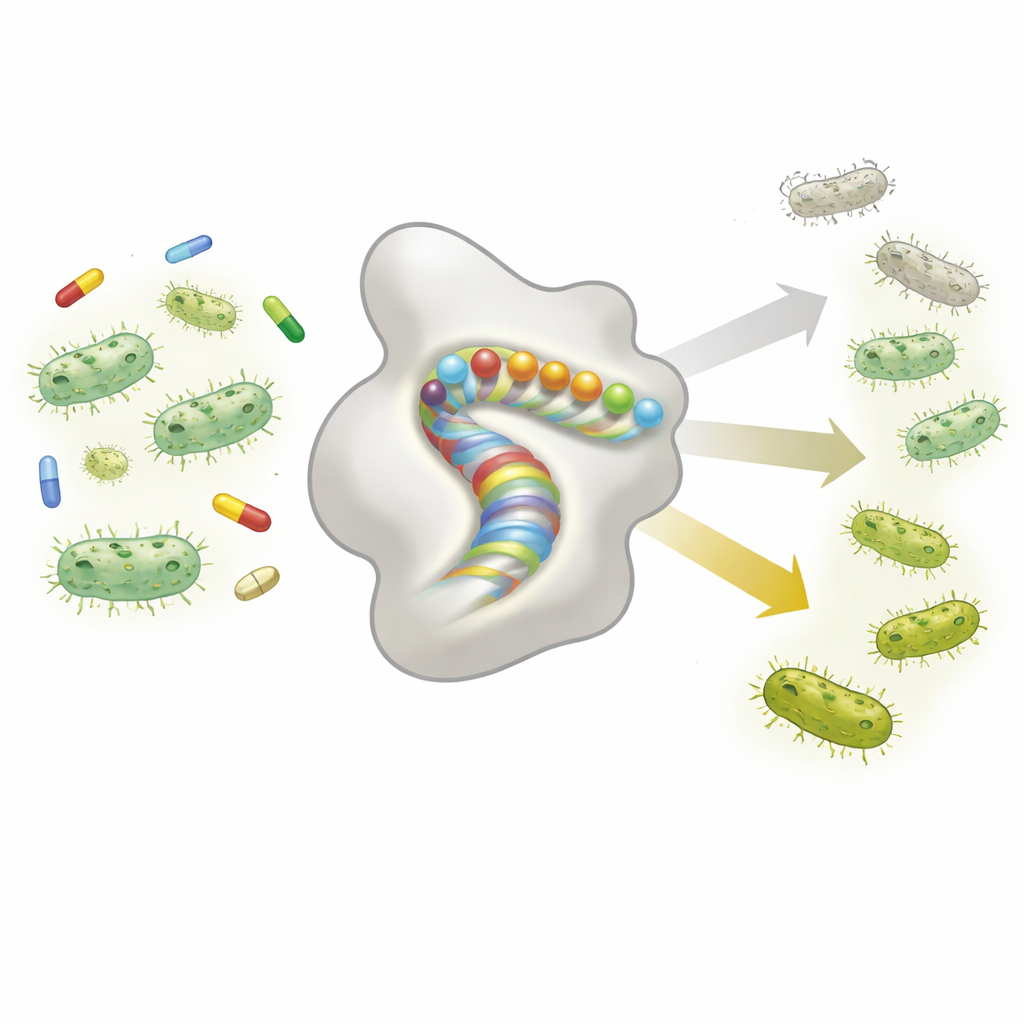
Cuando una mutación depende de otra
La idea central explorada aquí es la “epistasia”, que significa que el efecto de una mutación depende de qué otras mutaciones están presentes. En vez de actuar de forma independiente, las mutaciones pueden ayudarse o perjudicarse entre sí de maneras sorprendentes. Los autores construyeron una enorme biblioteca de más de 14.000 variantes de la enzima TEM-1, cada una con uno o dos cambios dentro de una única α-hélice que no forma parte del sitio activo pero contribuye a mantener la forma de la proteína. Luego midieron cuánto protegía cada variante a las bacterias frente a la amoxicilina de dos maneras: siguiendo el crecimiento bacteriano en competencia (una medida directa de la aptitud) y determinando la concentración mínima de antibiótico que impide el crecimiento (la CIM o MIC, una medida clínica estándar de resistencia). Estas mediciones emparejadas les permitieron cuantificar con qué frecuencia los efectos de las mutaciones dependen del contexto en lugar de ser simplemente aditivos.
Muchas mutaciones destruyen la enzima, pero la mayoría de interacciones son previsibles
El primer resultado llamativo es lo frágil que es esta pequeña hélice. Casi la mitad de todas las mutaciones simples en la región destruyeron esencialmente la función de la enzima en las condiciones de prueba, y casi cuatro de cada cinco dobles fueron no funcionales. Sustituciones concretas, como introducir la prolina, fueron casi siempre letales porque alteran la geometría de la hélice. Cuando el equipo comparó el comportamiento de los dobles con lo que se esperaría sumando los efectos de cada mutación simple, encontraron epistasia generalizada: una vez excluidas las variantes verdaderamente inactivas, alrededor del 90% de las mutaciones mostraron una dependencia clara del contexto. Sin embargo, la mayor parte de esta epistasia siguió un patrón simple: las mutaciones tendían a resultar peores sobre fondos ya debilitados, una forma de interacción negativa “global”.
Un modelo simple de plegamiento explica la mayor parte de la complejidad
Para entender por qué surgen estos patrones, los autores recurrieron a un modelo clásico de plegamiento en dos estados. En esta visión, TEM-1 está plegada y funcional o desplegada e inútil, y cada mutación desplaza el equilibrio entre estos estados al aumentar o disminuir la estabilidad de la proteína. Crucialmente, el modelo asume que los cambios de estabilidad de mutaciones individuales se suman, aunque el efecto final sobre la aptitud es no lineal: una vez que la proteína es lo bastante inestable, pequeños cambios adicionales tienen consecuencias mucho mayores. Al ajustar este marco a sus datos, los investigadores pudieron asignar a cada mutación un “coste de estabilidad” o “ganancia de estabilidad” efectivos. Con solo estos valores y un único parámetro de estabilidad de referencia, el modelo reprodujo de forma aproximada tanto la aptitud medida como la MIC de miles de mutantes simples y dobles, así como la distribución y el sesgo global de las interacciones epistáticas. En otras palabras, gran parte de la aparente complejidad de las interacciones mutacionales surge de una restricción física simple: la necesidad de que la proteína permanezca plegada.
Los contactos locales y la evolución a largo plazo dejan una huella más sutil
La historia no es puramente global, sin embargo. El modelo de dos estados funcionó menos bien para pares de residuos que están muy próximos en la estructura 3D de la enzima. Para esos vecinos, los datos revelaron epistasia “idiosincrática”: combinaciones específicas que se comportaron mejor o peor de lo esperado solo por la estabilidad, a veces compensándose mutuamente en carga o forma. Para ver si la evolución había preservado firmas de estas interacciones locales, los autores analizaron miles de secuencias de β-lactamasas relacionadas usando un modelo estadístico “Potts” que capta qué residuos tienden a variar juntos entre especies. Estos acoplamientos basados en secuencias se alinearon de dos maneras con los hallazgos experimentales: se correlacionaron con los efectos de estabilidad inferidos y destacaron muchos de los mismos pares de residuos donde el modelo de plegamiento simple fallaba. Cuando las salidas del modelo Potts se reescalaron cuidadosamente y se integraron en el mismo marco de plegamiento, incluso pudieron predecir en parte el signo de la epistasia para nuevos pares de mutaciones.
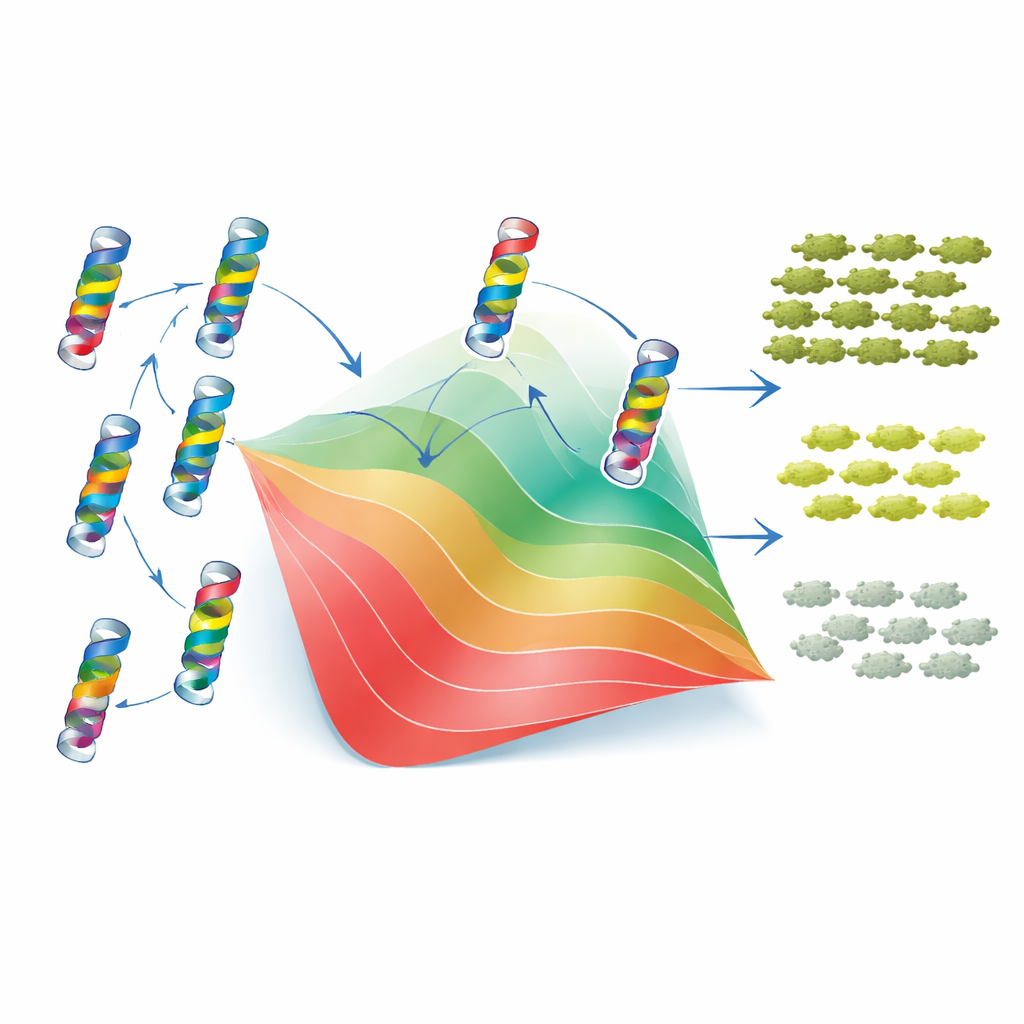
Qué significa esto para predecir la resistencia
Para quienes no son especialistas, la conclusión clave es que incluso en un elemento estructural diminuto de una sola proteína, las interacciones entre mutaciones son comunes pero no aleatorias. La mayoría pueden entenderse como consecuencias de cómo las mutaciones empujan colectivamente a la enzima hacia o lejos de un estado plegado estable, mientras que una minoría refleja contactos locales directos que afinan el comportamiento. El hecho de que ambos tipos de efectos dejen huellas detectables en la evolución a largo plazo de las β-lactamasas sugiere que las reglas de interacción mutacional son lo suficientemente estables como para aprenderse y generalizarse. Esto abre la posibilidad de que, combinando escaneos mutacionales profundos con modelos de plegamiento proteico y datos evolutivos de secuencias, eventualmente podamos prever cómo cambiarán las enzimas de resistencia —y diseñar fármacos o terapias que les resulte más difícil sortearlos.
Cita: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Palabras clave: epistasia, estabilidad proteica, beta-lactamasa, resistencia a antibióticos, escaneo mutacional profundo