Clear Sky Science · pt
Origens e abrangência da epistasia par a par em um α-hélice da β-lactamase TEM-1
Por que pequenas mudanças em uma proteína importam para a resistência a antibióticos
Bactérias resistentes a antibióticos representam uma ameaça crescente, e muitas delas dependem de enzimas que degradam os fármacos antes que estes possam agir. Uma dessas enzimas, chamada β-lactamase TEM-1, ajuda bactérias intestinais comuns a resistir a antibióticos amplamente usados, como a amoxicilina. Este estudo foca em uma porção muito pequena dessa enzima — uma espiral de 11 aminoácidos chamada α-hélice — para fazer uma grande pergunta: como pares de mutações se combinam para facilitar ou dificultar a evolução da resistência, e podemos prever esses efeitos a partir de princípios físicos básicos e de dados evolutivos?
Quando uma mutação depende de outra
A ideia central explorada aqui é a “epistasia”, que significa que o efeito de uma mutação depende de quais outras mutações estão presentes. Em vez de agir de forma independente, mutações podem se ajudar ou se atrapalhar de maneiras inesperadas. Os autores construíram uma grande biblioteca com mais de 14.000 variantes da enzima TEM-1, cada uma carregando uma ou duas alterações dentro de uma única α-hélice que não faz parte do sítio ativo, mas ajuda a manter a forma da proteína. Em seguida, mediram quão bem cada variante protegia bactérias contra a amoxicilina de duas maneiras: acompanhando o crescimento das bactérias em competição (uma medida direta de aptidão) e determinando a concentração mínima de antibiótico que impede o crescimento (MIC, uma medida clínica padrão de resistência). Essas medições pareadas permitiram quantificar com que frequência os efeitos das mutações são dependentes do contexto em vez de simplesmente aditivos.
Muitas mutações quebram a enzima, mas a maioria das interações é previsível
O primeiro resultado impressionante é quão frágil essa pequena hélice é. Quase metade de todas as mutações simples na região essencialmente destruíram a função da enzima nas condições do experimento, e quase quatro em cada cinco duplos mutantes foram não funcionais. Certas substituições, como a introdução do aminoácido prolina, foram quase sempre letais porque perturbam a conformação da hélice. Quando a equipe examinou como os duplos mutantes se comportavam em comparação com o que seria esperado pela soma dos efeitos de cada mutação simples, encontraram epistasia difundida: excluindo variantes realmente inativas, cerca de 90% das mutações mostraram dependência clara do contexto. No entanto, grande parte dessa epistasia segue um padrão simples — mutações tendiam a se comportar pior em antecedentes já enfraquecidos, uma forma de interação negativa “global”.
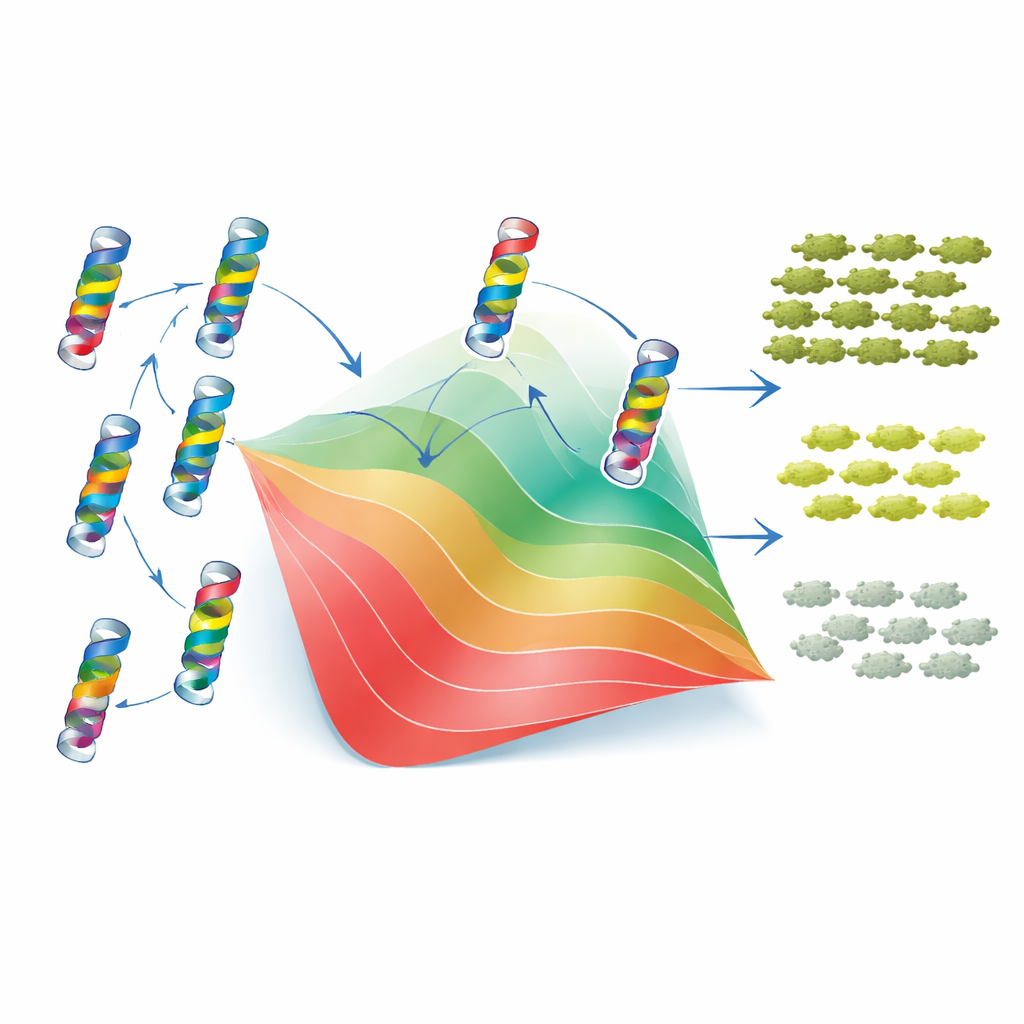
Um modelo simples de dobramento explica a maior parte da complexidade
Para entender por que esses padrões surgem, os autores recorreram a um clássico modelo de dobramento de duas fases. Nessa visão, a TEM-1 está ou dobrada e funcional, ou desnaturada e inútil, e cada mutação desloca o equilíbrio entre esses estados ao aumentar ou diminuir a estabilidade da proteína. Crucialmente, o modelo assume que as mudanças de estabilidade provocadas por mutações individuais se somam, embora o efeito final sobre a aptidão seja não linear: uma vez que a proteína fica instável o suficiente, pequenas alterações adicionais têm consequências muito maiores. Ao ajustar esse arcabouço aos seus dados, os pesquisadores puderam atribuir a cada mutação um “custo de estabilidade” ou “ganho de estabilidade” efetivo. Com apenas esses valores e um único parâmetro de estabilidade basal, o modelo reproduziu de forma aproximada tanto a aptidão medida quanto o MIC de milhares de mutantes simples e duplos, assim como a dispersão e o viés geral das interações epistáticas. Em outras palavras, grande parte da complexidade aparente das interações entre mutações decorre de uma restrição física simples: a necessidade da proteína permanecer dobrada.
Contatos locais e a evolução de longo prazo deixam uma marca mais sutil
A história não é puramente global, porém. O modelo de duas fases funcionou pior para pares de resíduos que ficam muito próximos na estrutura 3D da enzima. Para esses vizinhos, os dados revelaram epistasia “idiossincrática” — combinações específicas que se comportaram melhor ou pior do que o esperado apenas a partir da estabilidade, às vezes se compensando por carga ou forma. Para verificar se a evolução preservou assinaturas dessas interações locais, os autores analisaram milhares de sequências relacionadas de β-lactamases usando um modelo estatístico “Potts” que captura quais resíduos tendem a variar juntos entre espécies. Esses acoplamentos baseados em sequência se alinharam de duas formas com os achados experimentais: eles se correlacionaram com os efeitos de estabilidade inferidos e destacaram muitos dos mesmos pares de resíduos onde o modelo simples de dobramento falhava. Quando as saídas do modelo Potts foram cuidadosamente reescaladas e colocadas através do mesmo arcabouço de dobramento, elas puderam até prever em parte o sinal da epistasia para novos pares de mutações.
O que isso significa para prever resistência
Para não especialistas, a conclusão principal é que, mesmo em um elemento estrutural minúsculo de uma única proteína, as interações entre mutações são comuns, mas não aleatórias. A maioria pode ser entendida como consequência de como as mutações, coletivamente, empurram a enzima em direção a um estado dobrado estável ou para longe dele, enquanto uma minoria reflete contatos locais diretos que afinam o comportamento. O fato de ambos os tipos de efeito deixarem rastros detectáveis na evolução de longo prazo das β-lactamases sugere que as regras de interação mutacional são estáveis o suficiente para serem aprendidas e generalizadas. Isso levanta a possibilidade de que, ao combinar varreduras mutacionais profundas com modelos de dobramento de proteínas e dados de sequência evolutiva, possamos eventualmente prever como enzimas de resistência vão mudar — e projetar fármacos ou terapias que sejam mais difíceis de serem vencidos por elas.
Citação: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Palavras-chave: epistasia, estabilidade de proteínas, beta-lactamase, resistência a antibióticos, varredura mutacional profunda