Clear Sky Science · en
Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1
Why tiny changes in a protein matter for antibiotic resistance
Antibiotic-resistant bacteria are a growing threat, and many of them rely on enzymes that break down drugs before they can work. One such enzyme, called TEM-1 β-lactamase, helps common gut bacteria resist widely used antibiotics like amoxicillin. This study zooms in on a very small part of that enzyme—an 11–amino acid spiral called an α-helix—to ask a big question: how do pairs of mutations combine to make resistance easier or harder to evolve, and can we predict these effects from basic physical principles and from evolutionary data?
When one mutation depends on another
The core idea explored here is “epistasis,” which means that the effect of one mutation depends on which other mutations are present. Instead of acting independently, mutations can help or hurt each other in surprising ways. The authors built a huge library of more than 14,000 variants of the TEM-1 enzyme, each carrying either one or two changes within a single α-helix that is not part of the active site but helps keep the protein’s shape. They then measured how well each variant protected bacteria from amoxicillin in two ways: by tracking the bacteria’s growth in competition (a direct measure of fitness) and by determining the minimum antibiotic concentration that prevents growth (the MIC, a standard clinical measure of resistance). These paired measurements allowed them to quantify how often mutation effects are context-dependent rather than simply additive.
Many mutations break the enzyme, but most interactions are predictable
The first striking result is how fragile this small helix is. Nearly half of all single mutations in the region essentially destroyed the enzyme’s function under the test conditions, and almost four out of five double mutants were non-functional. Certain substitutions, such as introducing the amino acid proline, were almost always lethal because they disrupt the helix’s shape. When the team examined how double mutants behaved compared with what would be expected from simply adding the effects of each single mutation, they found widespread epistasis: once truly dead variants were excluded, about 90% of mutations showed clear context dependence. However, most of this epistasis had a simple pattern—mutations tended to look worse on already weakened backgrounds, a form of “global” negative interaction.
A simple folding model explains most of the complexity
To understand why these patterns arise, the authors turned to a classic two-state model of protein folding. In this view, TEM-1 is either folded and functional or unfolded and useless, and each mutation shifts the balance between these states by raising or lowering the protein’s stability. Crucially, the model assumes that the stability changes from individual mutations add up, even though the final effect on fitness is non-linear: once the protein is unstable enough, small extra changes have much larger consequences. By fitting this framework to their data, the researchers could assign each mutation an effective “stability cost” or “stability gain.” With just these values and a single baseline stability parameter, the model closely reproduced both the measured fitness and MIC of thousands of single and double mutants, as well as the overall spread and bias of epistatic interactions. In other words, much of the apparent complexity of mutation interactions arises from a simple physical constraint: the need for the protein to stay folded.
Local contacts and long-term evolution leave a subtler mark
The story is not purely global, however. The two-state model worked less well for pairs of residues that sit very close together in the 3D structure of the enzyme. For such neighbors, the data revealed “idiosyncratic” epistasis—specific combinations that behaved better or worse than expected from stability alone, sometimes compensating for each other’s charge or shape. To see whether evolution had preserved signatures of these local interactions, the authors analyzed thousands of related β-lactamase sequences using a statistical “Potts” model that captures which residues tend to vary together across species. These sequence-based couplings lined up in two ways with the experimental findings: they correlated with the inferred stability effects and highlighted many of the same residue pairs where the simple folding model failed. When the Potts model’s outputs were carefully rescaled and fed through the same folding framework, they could even partly predict the sign of epistasis for new mutation pairs.
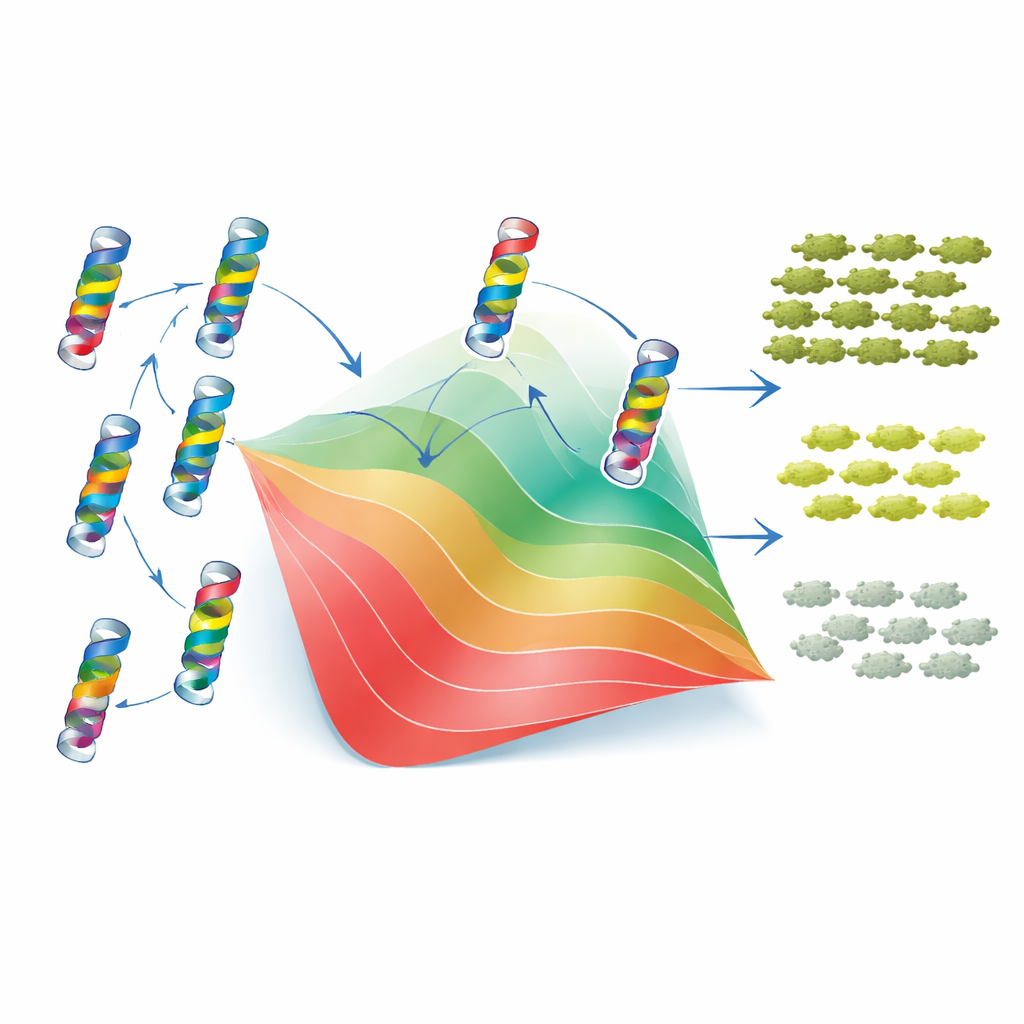
What this means for predicting resistance
For non-specialists, the key takeaway is that even in a tiny structural element of a single protein, interactions between mutations are common but not random. Most can be understood as consequences of how mutations collectively push the enzyme toward or away from a stable folded state, while a minority reflect direct local contacts that fine-tune behavior. The fact that both types of effects leave detectable traces in the long-term evolution of β-lactamases suggests that the rules of mutational interaction are stable enough to be learned and generalized. This raises the possibility that, by combining deep mutational scans with models of protein folding and evolutionary sequence data, we may eventually be able to forecast how resistance enzymes will change—and to design drugs or therapies that are harder for them to outsmart.
Citation: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Keywords: epistasis, protein stability, beta-lactamase, antibiotic resistance, deep mutational scanning