Clear Sky Science · de
Ursprung und Ausmaß paarweiser Epistasen in einer α‑Helix von β‑Lactamase TEM‑1
Warum winzige Veränderungen in einem Protein für Antibiotikaresistenz wichtig sind
Antibiotikaresistente Bakterien sind eine wachsende Bedrohung, und viele von ihnen nutzen Enzyme, die Medikamente abbauen, bevor diese wirken können. Ein solches Enzym, TEM‑1 β‑Lactamase genannt, hilft häufigen Darmbakterien, gegen weit verbreitete Antibiotika wie Amoxicillin resistent zu sein. Diese Studie richtet den Blick auf einen sehr kleinen Teil dieses Enzyms — eine 11‑Aminosäuren‑Spirale, eine α‑Helix — und stellt eine große Frage: Wie kombinieren sich Paarmutationen, um die Evolution von Resistenz zu erleichtern oder zu erschweren, und lassen sich diese Effekte aus grundlegenden physikalischen Prinzipien und evolutiven Daten vorhersagen?
Wenn eine Mutation von einer anderen abhängt
Die hier untersuchte Kernidee ist „Epistasie“, also dass die Wirkung einer Mutation davon abhängt, welche anderen Mutationen vorhanden sind. Anstatt unabhängig zu wirken, können Mutationen einander in überraschender Weise begünstigen oder behindern. Die Autoren bauten eine riesige Bibliothek mit mehr als 14.000 Varianten des TEM‑1‑Enzyms auf, von denen jede entweder eine oder zwei Veränderungen innerhalb einer einzigen α‑Helix trägt — diese Helix ist nicht Teil des aktiven Zentrums, hilft aber, die Form des Proteins zu stabilisieren. Anschließend maßen sie, wie gut jede Variante die Bakterien vor Amoxicillin schützte, auf zwei Arten: durch das Wachstum der Bakterien im Wettbewerb (ein direkter Fitness‑Messwert) und durch Bestimmung der minimalen Antibiotikakonzentration, die Wachstum verhindert (MIC, ein klinischer Standardwert für Resistenz). Diese gepaarten Messungen erlaubten es, zu quantifizieren, wie häufig Mutationen kontextabhängig statt einfach additiv sind.
Viele Mutationen zerstören das Enzym, aber die meisten Interaktionen sind vorhersagbar
Das erste auffällige Ergebnis ist, wie zerbrechlich diese kleine Helix ist. Nahezu die Hälfte aller Einzelmutationen in der Region zerstörte unter den Testbedingungen im Wesentlichen die Enzymfunktion, und fast vier von fünf Doppelmutanten waren nicht funktional. Bestimmte Substitutionen, etwa die Einführung der Aminosäure Prolin, waren fast immer tödlich, weil sie die Helixstruktur stören. Verglich das Team das Verhalten der Doppelmutanten mit dem, was man durch simple Addition der Effekte der Einzelmutationen erwarten würde, so fand sich weit verbreitete Epistasie: Wurden wirklich tote Varianten ausgeschlossen, zeigten etwa 90 % der Mutationen eine klare Kontextabhängigkeit. Doch die meiste Epistasie folgte einem einfachen Muster — Mutationen wirkten tendenziell schlechter auf bereits geschwächten Hintergründen, eine Form von „globaler“ negativer Wechselwirkung.
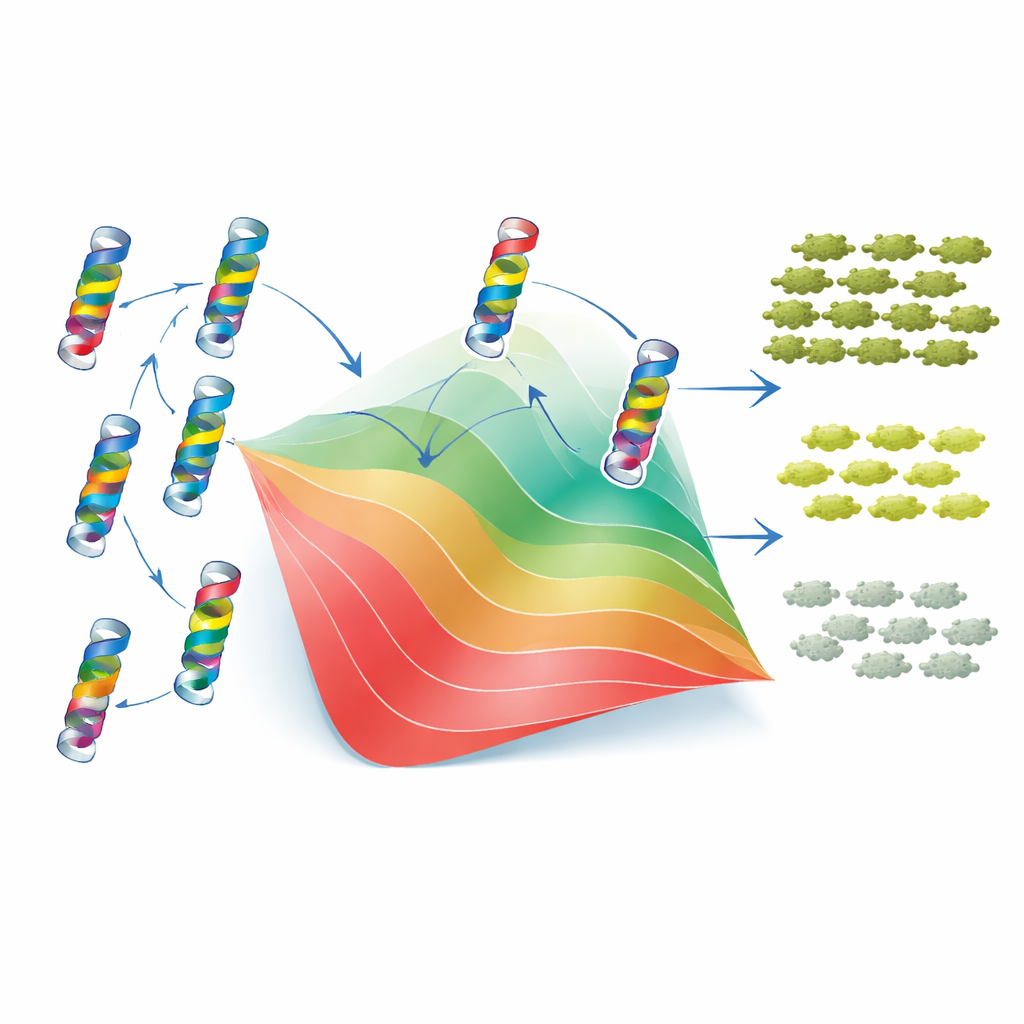
Ein einfaches Faltungsmodell erklärt den Großteil der Komplexität
Um zu verstehen, warum diese Muster entstehen, griffen die Autoren auf ein klassisches Zwei‑Zustands‑Modell der Proteinfaltung zurück. Nach dieser Sichtweise ist TEM‑1 entweder gefaltet und funktionell oder ungefaltet und nutzlos, und jede Mutation verschiebt das Gleichgewicht zwischen diesen Zuständen, indem sie die Stabilität des Proteins hebt oder senkt. Entscheidend ist, dass das Modell annimmt, die Stabilitätsänderungen einzelner Mutationen addierten sich, obwohl der endgültige Effekt auf die Fitness nichtlinear ist: Sobald das Protein instabil genug ist, haben kleine zusätzliche Veränderungen deutlich größere Konsequenzen. Durch Anpassung dieses Rahmens an ihre Daten konnten die Forschenden jeder Mutation einen effektiven „Stabilitätskosten“- oder „Stabilitätsgewinn“-Wert zuweisen. Mit nur diesen Werten und einem einzigen Basis‑Stabilitätsparameter reproduzierte das Modell sehr gut sowohl die gemessene Fitness als auch die MIC‑Werte von Tausenden Einzel‑ und Doppelmutanten sowie die allgemeine Verteilung und Verzerrung epistatischer Wechselwirkungen. Anders gesagt: Ein Großteil der scheinbaren Komplexität von Mutationswechselwirkungen ergibt sich aus einer einfachen physikalischen Einschränkung — der Notwendigkeit, dass das Protein gefaltet bleibt.
Lokale Kontakte und langfristige Evolution hinterlassen subtilere Spuren
Die Geschichte ist jedoch nicht rein global. Das Zwei‑Zustands‑Modell funktionierte weniger gut für Paare von Resten, die im 3D‑Strukturmodell des Enzyms sehr nahe beieinander liegen. Bei solchen Nachbarn zeigten die Daten „idiosynkratische“ Epistasen — spezifische Kombinationen, die besser oder schlechter reagierten als durch Stabilität allein vorhergesagt, wobei sie sich manchmal gegenseitig in Ladung oder Form kompensierten. Um zu prüfen, ob die Evolution Signaturen dieser lokalen Interaktionen bewahrt hat, analysierten die Autoren Tausende verwandter β‑Lactamase‑Sequenzen mit einem statistischen „Potts“‑Modell, das erfasst, welche Reste dazu neigen, über Arten hinweg zusammen zu variieren. Diese sequenzbasierten Kopplungen stimmten auf zweierlei Weise mit den experimentellen Befunden überein: Sie korrelierten mit den abgeleiteten Stabilitätseffekten und hoben viele der gleichen Restpaare hervor, bei denen das einfache Faltungsmodell versagte. Wenn die Ausgaben des Potts‑Modells sorgfältig skaliert und durch denselben Faltungsrahmen geschickt wurden, konnten sie sogar teilweise das Vorzeichen der Epistasie für neue Mutationspaare vorhersagen.
Was das für die Vorhersage von Resistenz bedeutet
Für Nicht‑Spezialisten ist die wichtigste Schlussfolgerung, dass selbst in einem winzigen Strukturelement eines einzelnen Proteins Interaktionen zwischen Mutationen häufig, aber nicht zufällig sind. Die meisten lassen sich als Folge dessen verstehen, wie Mutationen gemeinsam das Enzym in Richtung eines stabilen, gefalteten Zustands drängen oder davon wegschieben, während eine Minderheit direkte lokale Kontakte widerspiegelt, die das Verhalten feinjustieren. Die Tatsache, dass beide Effektarten in der langfristigen Evolution von β‑Lactamasen nachweisbare Spuren hinterlassen, legt nahe, dass die Regeln der mutationalen Interaktion stabil genug sind, um erlernt und verallgemeinert zu werden. Das eröffnet die Möglichkeit, dass sich durch die Kombination von Deep Mutational Scans mit Modellen der Proteinfaltung und evolutionären Sequenzdaten künftig vorhersagen lässt, wie Resistenzenzyme sich verändern werden — und dass man Medikamente oder Therapien entwerfen kann, denen sie schwerer ausweichen können.
Zitation: Birgy, A., Roussel, C., Kemble, H. et al. Origins and breadth of pairwise epistasis in an α-helix of β-lactamase TEM-1. Nat Commun 17, 4083 (2026). https://doi.org/10.1038/s41467-026-70627-5
Schlüsselwörter: Epistasie, Proteinstabilität, Beta‑Lactamase, Antibiotikaresistenz, Deep Mutational Scanning