Clear Sky Science · ru
Таргетное длинно-ридовое секвенирование для высокоразрешающего профилирования повторов при миотонической дистрофии 1 типа
Почему это важно для семей и врачей
Некоторые наследственные заболевания вызваны «заиканиями» в нашей ДНК — короткими последовательностями, повторяющимися сотни или даже тысячи раз. Когда эти повторы слишком удлиняются, они могут вызвать тяжёлые состояния, такие как миотоническая дистрофия, заболевание, приводящее к истощению мышц. Тем не менее точное измерение длины таких повторов и их химической маркировки оказывается неожиданно сложной задачей для современных лабораторных тестов. В этой статье представлен упрощённый подход к чтению этих трудных участков ДНК с высоким разрешением примерно за сутки, что потенциально даёт врачам более ясные ответы, а пациентам — лучшее руководство.
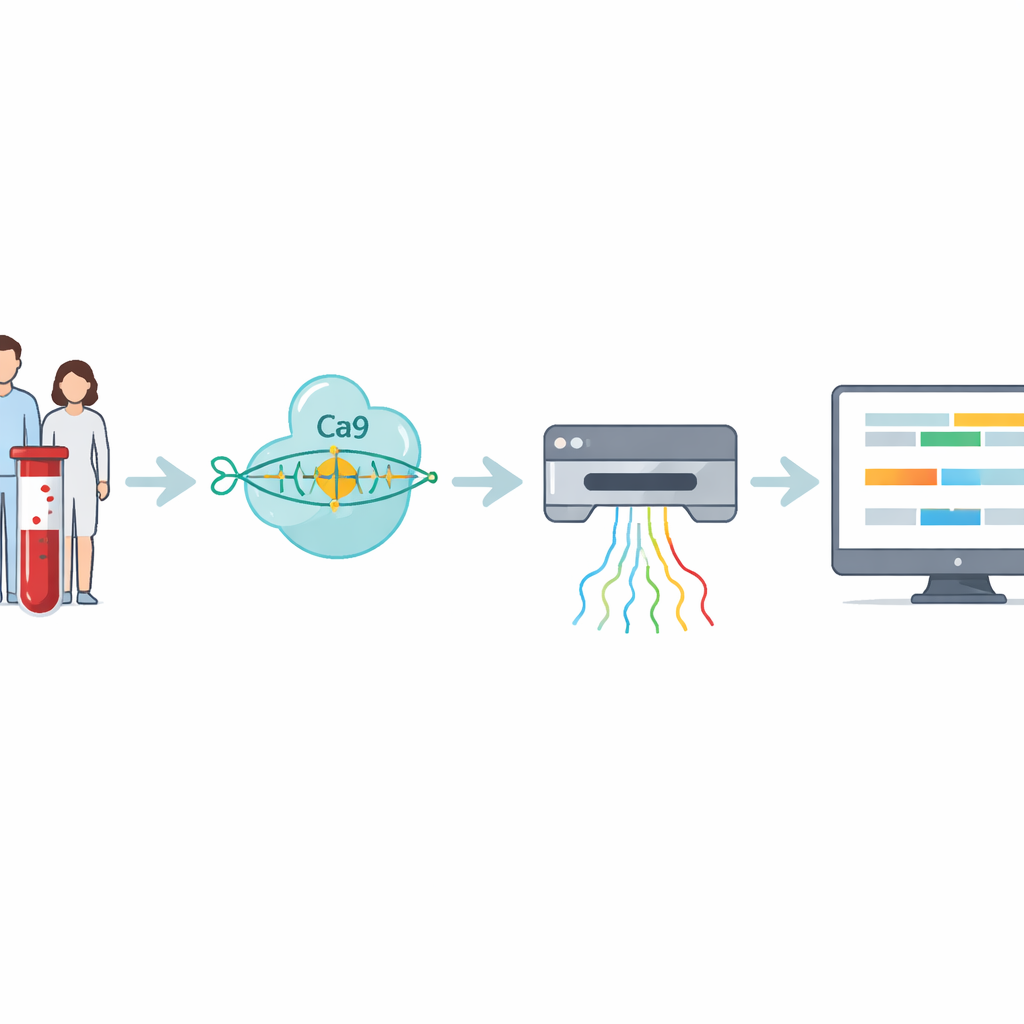
Более пристальный взгляд на сложное мышечное заболевание
Миотоническая дистрофия типа 1 (DM1) вызвана чрезмерным удлинением триплетной последовательности в гене, важном для мышц и других тканей. У людей с большим числом повторов заболевание, как правило, более тяжёлое, поэтому знание точной длины повтора важно для диагностики и прогноза. Стандартные методы в клинических лабораториях — такие как специализированный ПЦР и южные блотты — хорошо работают, когда размеры повторов умеренные, но часто терпят неудачу или дают очень неточные результаты, если экспансии превышают несколько сотен копий. Это оставляет «серую зону», когда семьи могут знать о наличии мутации, но не иметь чёткого представления о её масштабе или о том, как она может изменяться со временем.
Сочетание молекулярных «ножниц» и длинного чтения ДНК
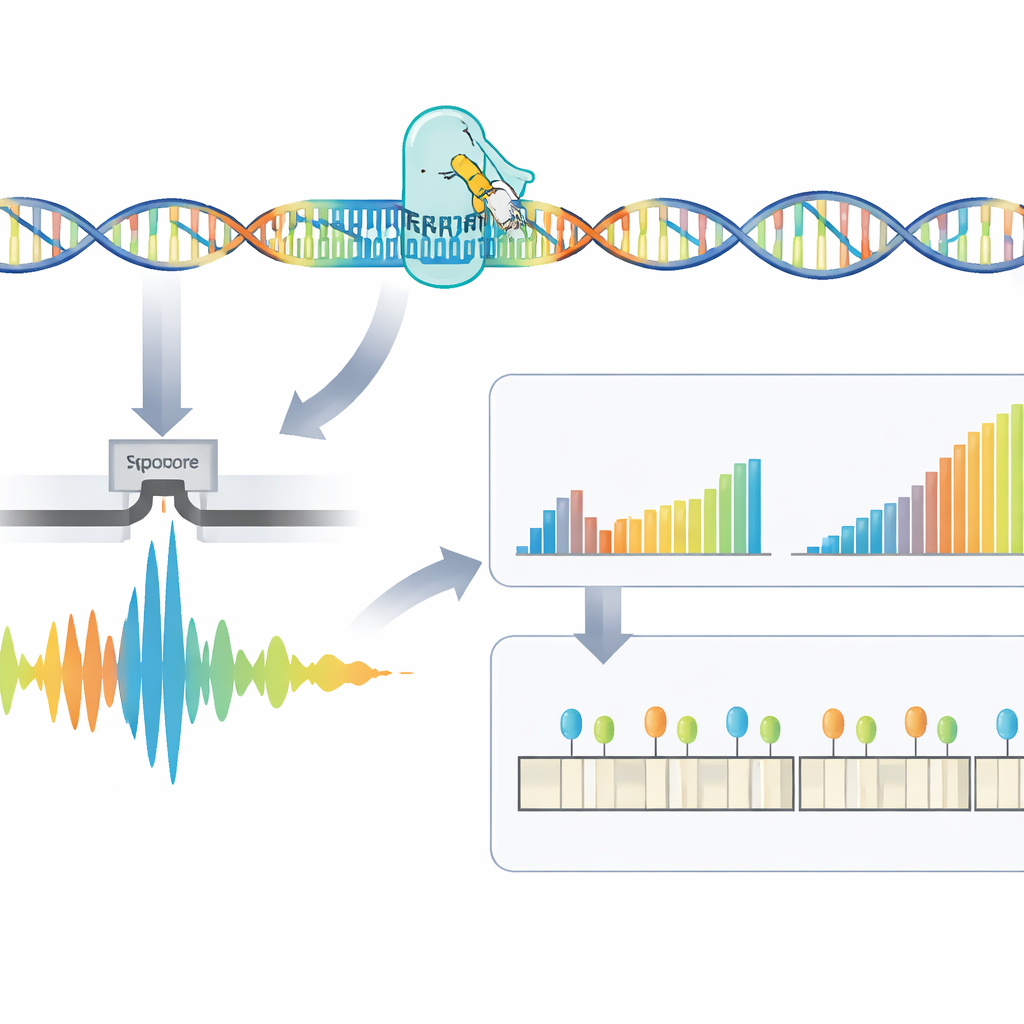
Авторы разработали четырёхэтапный рабочий процесс, который напрямую справляется с этими трудночитаемыми повторами. Сначала ДНК извлекают из крови или клеточных образцов. Затем группа использует CRISPR–Cas9 — молекулярные «ножницы», направляемые к определённому участку генома — чтобы вырезать фрагменты вокруг участка с повтором. Поскольку для секвенирования готовят только эти вырезанные фрагменты, метод сильно обогащает интересующий регион, избегая амплификации ДНК методом ПЦР, которая обычно пропускает очень крупные экспансии. Подготовленную ДНК затем загружают в нанопоровый секвенатор — портативное устройство, которое протаскивает молекулу ДНК через маленькие поры и в реальном времени фиксирует электрические сигналы, отражающие её состав.
Умное ПО для определения длины и химических меток
Сырые сигналы от секвенатора нужно преобразовать в код ДНК и затем в содержательные метрики. Здесь авторы представляют RepeatLab, автоматизированный аналитический конвейер, рассчитанный на работу даже на обычном облачном ноутбуке. RepeatLab сначала быстро отбирает риды, покрывающие целевой ген, а затем повторно обрабатывает эти риды в более требовательном, высокоточном режиме, адаптированном для долгих повторяющихся участков. Он использует модифицированную статистическую стратегию для кластеризации измерений отдельных молекул в две группы — обычно нормальную и расширенную аллели у каждого человека — обеспечивая надёжные оценки длины уже при нескольких десятках ридов. Та же платформа может анализировать множество образцов или несколько генов в одном запуске, сохраняя затраты сопоставимыми с существующими клиническими методиками.
Взгляд на прерывания и эпигенетические сигналы
Помимо простого подсчёта повторов, метод исследует тонкую структуру повторяющегося региона и его химическую «отделку». У некоторых пациентов внутри повтора встречаются прерывания — небольшие участки с иной последовательностью, которые могут быть связаны с более мягким клиническим проявлением. Авторы обнаружили, что многие кажущиеся прерывания на самом деле были артефактами обработки сырого сигнала, и показали, что применение больших аналитических окон при декодировании значительно уменьшает количество ложноположительных находок. RepeatLab также включает специализированный режим чтения метилирования ДНК — химической метки, способной переключать активность генов. Вокруг гена DM1 исследователи картировали паттерны метилирования с разрешением по отдельным аллелям и выделили несколько ключевых областей, где более интенсивное метилирование коррелирует с более длинными экспансиями повторов, что поддерживает идею о том, что эти химические метки влияют на развитие болезни.
Быстрый и информативный тест для трудноизмеримых повторов
В совокупности работа демонстрирует, что обогащение с помощью направленного CRISPR, нанопоровое длинно-ридовое секвенирование и удобное в использовании программное обеспечение могут предоставить детализированный профиль патогенных ДНК-повторов менее чем за 24 часа. Для DM1 и родственных состояний подход не только измеряет длину повторов, но и фиксирует тонкие изменения последовательности и эпигенетические паттерны, которые в значительной степени ускользают от текущих клинических тестов. Хотя необходимы дополнительные валидация и адаптация к новым секвенирующим химиям, эта интегрированная платформа указывает путь к будущим генетическим тестам, которые будут быстрее, информативнее и проще в повседневном использовании.
Цитирование: Han, Y., Jang, JH. & Chang, H. Targeted long-read sequencing for high-resolution repeat profiling in myotonic dystrophy type 1. Exp Mol Med 58, 1203–1215 (2026). https://doi.org/10.1038/s12276-026-01683-6
Ключевые слова: миотоническая дистрофия, экспансия тандемных повторов, нанопоровое секвенирование, обогащение с помощью CRISPR Cas9, метилирование ДНК