Clear Sky Science · es
Secuenciación dirigida de lecturas largas para un perfil de alta resolución de repeticiones en distrofia miotónica tipo 1
Por qué importa esto para familias y médicos
Algunas enfermedades hereditarias son impulsadas por tramos repetitivos en nuestro ADN: secuencias cortas repetidas cientos o incluso miles de veces. Cuando estas repeticiones se alargan demasiado, pueden causar afecciones graves como la distrofia miotónica, una enfermedad que provoca pérdida de masa muscular. Sin embargo, medir con precisión la longitud de estas repeticiones y cómo están marcadas químicamente resulta sorprendentemente difícil con las pruebas hospitalarias actuales. Este artículo presenta una forma optimizada de leer en detalle estas regiones problemáticas del ADN en aproximadamente un día, lo que podría ofrecer a los médicos respuestas más claras y a los pacientes una mejor orientación.
Una mirada más cercana a una enfermedad muscular complicada
La distrofia miotónica tipo 1 (DM1) es causada por una repetición excesiva de tres letras del ADN en un gen importante para el músculo y otros tejidos. Las personas con más repeticiones tienden a presentar una enfermedad más grave, por lo que conocer la longitud exacta de la repetición es vital para el diagnóstico y el pronóstico. Las herramientas estándar en laboratorios clínicos—como ensayos PCR especializados y Southern blot—funcionan bien cuando la repetición es de tamaño moderado, pero a menudo fallan o se vuelven muy imprecisas cuando las expansiones superan unas pocas centenas de copias. Esto deja una zona gris en la que las familias pueden saber que existe una mutación pero carecer de una imagen clara sobre su extensión o su posible evolución en el tiempo.
Combinando tijeras moleculares y lectura larga del ADN
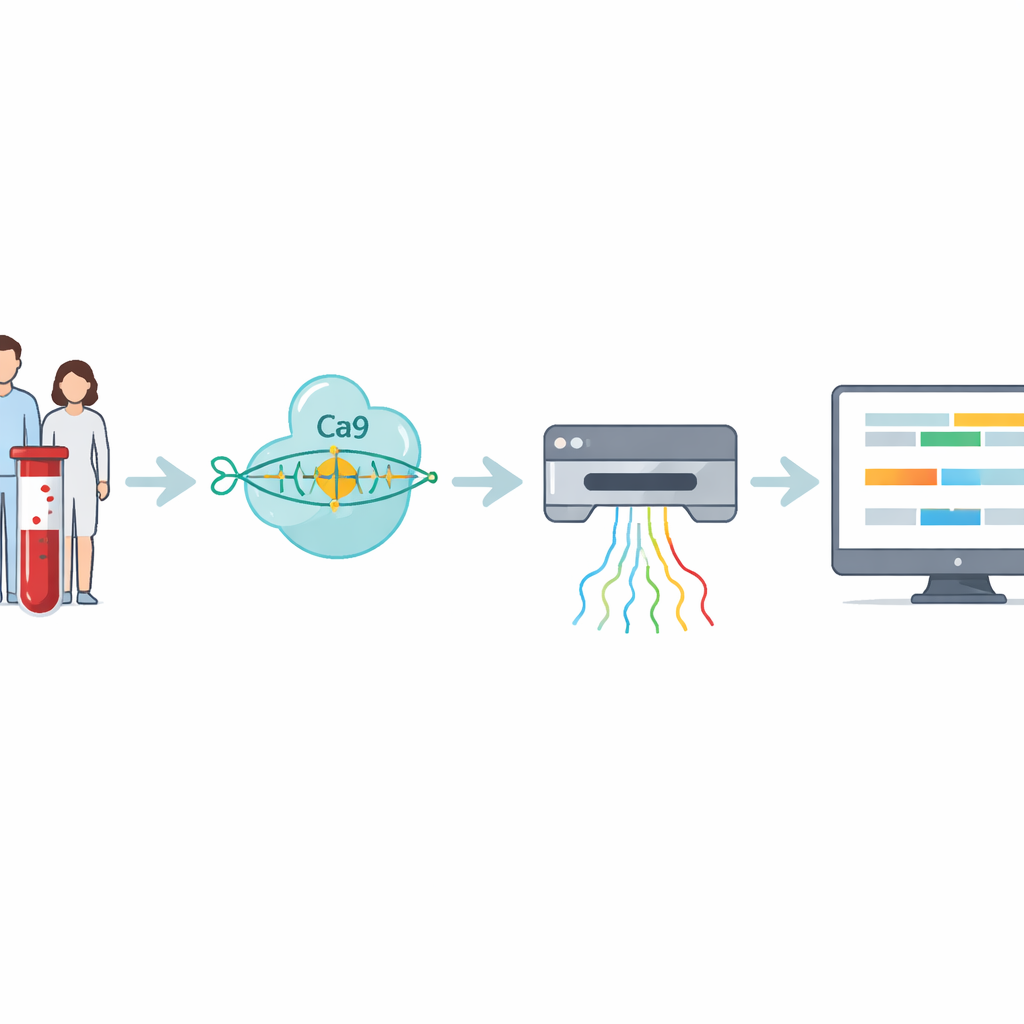
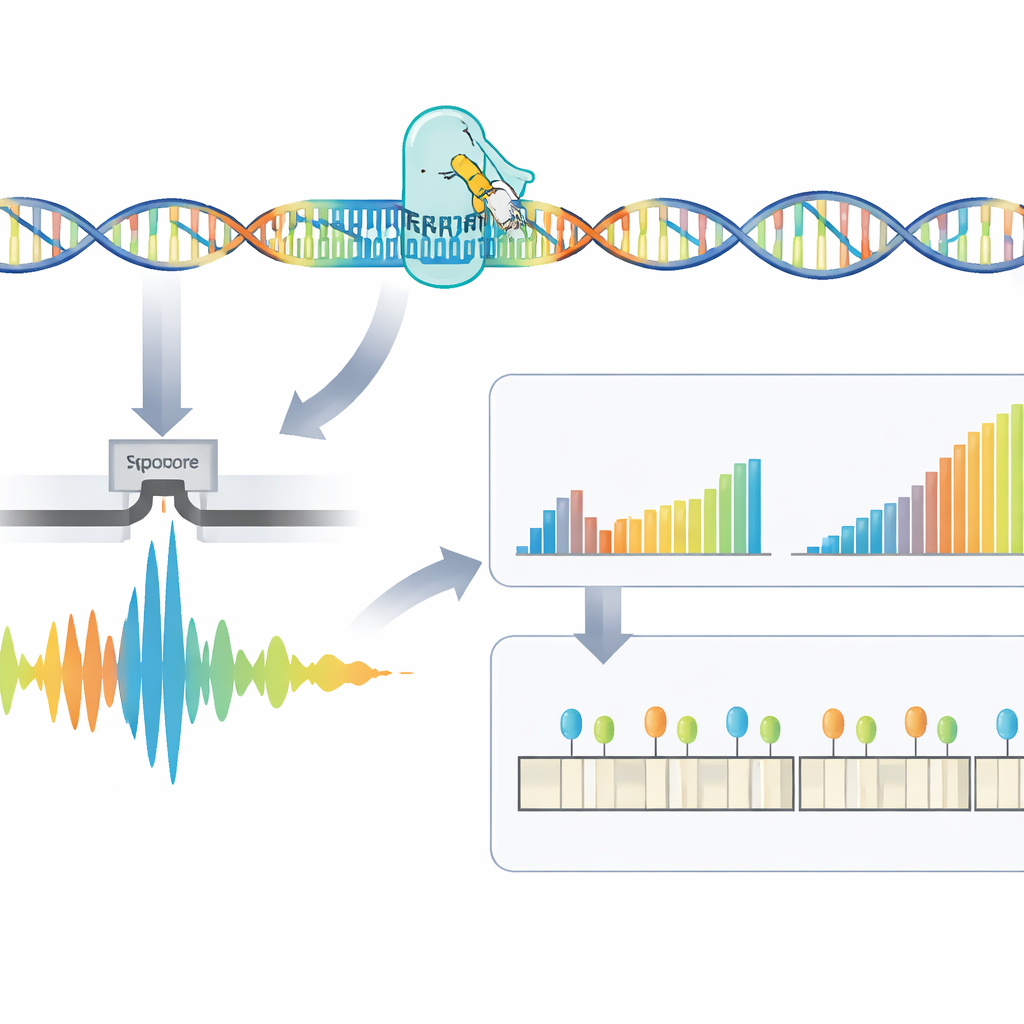
Los investigadores desarrollaron un flujo de trabajo de cuatro pasos que aborda directamente estas repeticiones difíciles de leer. Primero, se extrae el ADN de sangre o muestras celulares. A continuación, el equipo usa CRISPR–Cas9—tijeras moleculares guiadas a una dirección específica del genoma—para cortar alrededor del tramo de ADN que contiene la repetición. Como solo se preparan para secuenciar las piezas cortadas, el método enriquece fuertemente la región de interés evitando la amplificación por PCR, que tiende a fallar con expansiones muy grandes. El ADN preparado se introduce luego en un secuenciador por nanoporo, un dispositivo de mano que hace pasar hebras de ADN a través de poros minúsculos y detecta su composición en tiempo real mediante señales eléctricas.
Software inteligente para leer longitudes y marcas químicas
Las señales crudas del secuenciador deben traducirse a código de ADN y luego a medidas significativas. Aquí los autores presentan RepeatLab, una canalización de análisis automatizada diseñada para poder ejecutarse incluso en un cuaderno en la nube corriente. RepeatLab realiza primero una pasada rápida para encontrar lecturas que cubran el gen objetivo y después reprocesa esas lecturas con una configuración más exigente y de alta precisión, adaptada a tramos largos y repetitivos. Emplea una estrategia estadística modificada para agrupar mediciones de moléculas individuales en dos conjuntos—típicamente la copia normal y la expandida del gen en cada persona—logrando estimaciones de longitud fiables con apenas unas doce lecturas. El mismo marco puede analizar muchas muestras o varios genes en una sola corrida, manteniendo los costes comparables a los ensayos clínicos existentes.
Explorando interrupciones y señales epigenéticas
Más allá de simplemente contar repeticiones, el método examina la estructura fina de la región repetida y cómo está químicamente decorada. Algunos pacientes presentan interrupciones—pequeños fragmentos de secuencia distinta dentro de la repetición—que podrían asociarse a una enfermedad más leve. El equipo halló que muchas aparentes interrupciones eran en realidad artefactos del procesamiento de la señal cruda, y demostraron que usar ventanas de análisis mayores durante la decodificación reducía mucho estos falsos positivos. RepeatLab también emplea un modo especializado para leer la metilación del ADN, una marca química que puede activar o silenciar genes. Alrededor del gen implicado en DM1, los autores cartografiaron patrones de metilación a resolución de alelo individual e identificaron varias zonas clave donde una metilación más intensa se correlaciona con expansiones de repetición más largas, apoyando la idea de que estas marcas químicas contribuyen a moldear la evolución de la enfermedad.
Una prueba más rápida y rica para repeticiones difíciles de medir
En conjunto, este trabajo muestra que el enriquecimiento guiado por CRISPR, la secuenciación de lecturas largas por nanoporo y software accesible pueden ofrecer un perfil detallado de repeticiones de ADN causantes de enfermedad en menos de 24 horas. Para DM1 y afecciones relacionadas, el enfoque no solo mide la longitud de las repeticiones, sino que también captura cambios sutiles en la secuencia y patrones epigenéticos que las pruebas hospitalarias actuales en gran medida pasan por alto. Aunque todavía se requiere mayor validación y adaptación a nuevas químicas de secuenciación, esta plataforma integrada apunta hacia futuras pruebas genéticas más rápidas, más informativas y más fáciles de desplegar en la atención de rutina.
Cita: Han, Y., Jang, JH. & Chang, H. Targeted long-read sequencing for high-resolution repeat profiling in myotonic dystrophy type 1. Exp Mol Med 58, 1203–1215 (2026). https://doi.org/10.1038/s12276-026-01683-6
Palabras clave: distrofia miotónica, expansión de repeticiones en tándem, secuenciación por nanoporo, enriquecimiento CRISPR Cas9, metilación del ADN