Clear Sky Science · pl
Celowane sekwencjonowanie długich odczytów dla wysokorozdzielczego profilowania powtórzeń w dystrofii miotonicznej typu 1
Dlaczego to ma znaczenie dla rodzin i lekarzy
Niektóre choroby dziedziczne wynikają z „zacinających się” fragmentów naszego DNA — krótkich sekwencji powtarzanych setki, a nawet tysiące razy. Gdy te powtórzenia rosną zbyt długo, mogą wywoływać poważne schorzenia, takie jak dystrofia miotoniczna, choroba prowadząca do zaniku mięśni. Dokładne zmierzenie długości tych powtórzeń i ich chemicznych znaków jest jednak zadaniem trudnym dla rutynowych badań szpitalnych. Artykuł przedstawia uproszczoną metodę pozwalającą odczytać te trudne regiony DNA w szczegółach w ciągu około doby, co może dać lekarzom jaśniejsze odpowiedzi, a pacjentom lepsze wskazówki.
Bliższe spojrzenie na trudną do zdiagnozowania chorobę mięśni
Dystrofia miotoniczna typu 1 (DM1) jest spowodowana nadmiernym wydłużeniem tri-nukleotydowej sekwencji w genie istotnym dla mięśni i innych tkanek. Osoby z większą liczbą powtórzeń zwykle mają cięższy przebieg choroby, dlatego znajomość dokładnej długości powtórzeń ma kluczowe znaczenie dla diagnozy i prognozy. Standardowe metody w laboratoriach klinicznych — takie jak wyspecjalizowane testy PCR i hybrydyzacja Southern — działają dobrze przy umiarkowanej długości powtórzeń, ale często zawodzą lub stają się bardzo niedokładne, gdy ekspansje przekraczają kilkaset kopii. Powstaje wtedy szara strefa, w której rodziny wiedzą o obecności mutacji, lecz nie mają jasnego obrazu jej rozmiaru ani tego, jak może się zmieniać w czasie.
Połączenie molekularnych nożyczek i odczytów długich
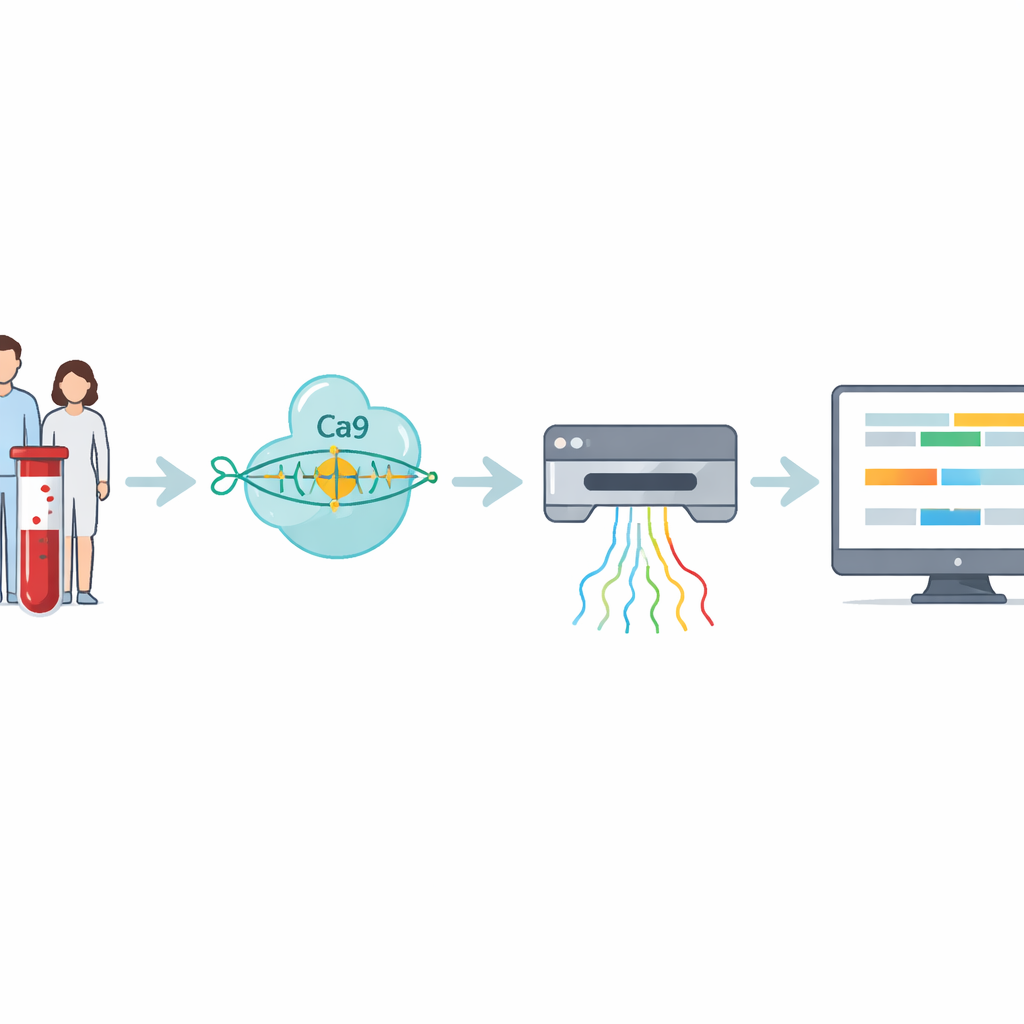
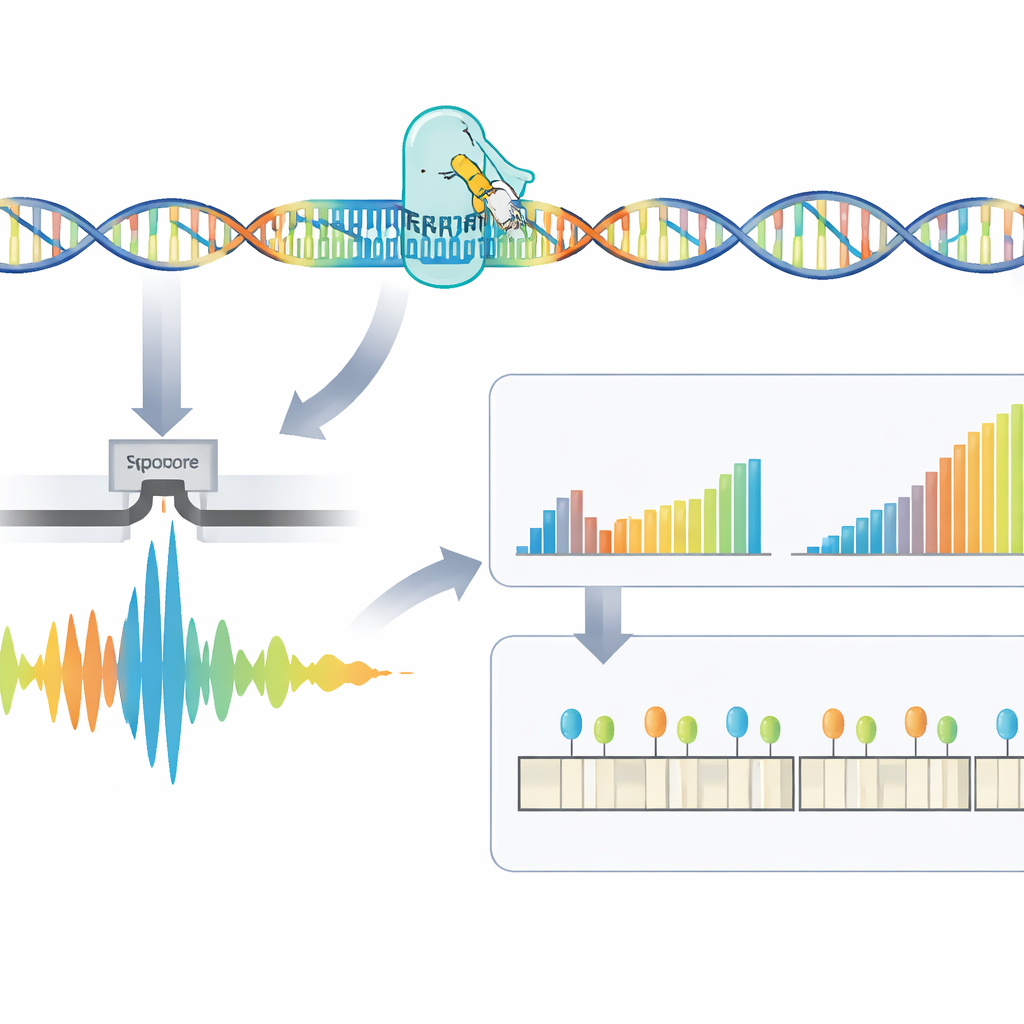
Naukowcy opracowali czterostopniowy workflow, który bezpośrednio radzi sobie z tymi trudnymi do odczytania powtórzeniami. Najpierw z próbki krwi lub komórek izoluje się DNA. Następnie zespół używa CRISPR–Cas9 — molekularnych nożyczek kierowanych do konkretnego miejsca w genomie — aby wyciąć fragment otaczający region z powtórzeniem. Ponieważ tylko te wycięte fragmenty są przygotowywane do sekwencjonowania, metoda silnie wzbogaca interesujący region, unikając amplifikacji PCR, która ma tendencję do pomijania bardzo dużych ekspansji. Przygotowane cząsteczki DNA trafiają do sekwenatora nanopore, przenośnego urządzenia, które przewleka nici DNA przez maleńkie pory i w czasie rzeczywistym rejestruje ich skład jako sygnały elektryczne.
Inteligentne oprogramowanie do określania długości i znaków chemicznych
Surowe sygnały z sekwenatora trzeba przetłumaczyć na kod DNA, a potem na użyteczne pomiary. Autorzy przedstawiają tu RepeatLab, zautomatyzowany pipeline analityczny zaprojektowany tak, by działać nawet na zwykłym notatniku w chmurze. RepeatLab najpierw wykonuje szybkie przeszukanie w celu znalezienia odczytów pokrywających celowany gen, a następnie ponownie przetwarza te odczyty przy użyciu bardziej wymagającego, wysokodokładnego ustawienia dostosowanego do długich, powtarzalnych fragmentów. Stosuje zmodyfikowaną strategię statystyczną do grupowania pomiarów pojedynczych cząsteczek w dwie klastry — zwykle kopię normalną i kopię z ekspansją u danej osoby — osiągając wiarygodne szacunki długości przy zaledwie kilkunastu odczytach. Ten sam system może analizować wiele próbek lub wiele genów w jednym runie, utrzymując koszty porównywalne z istniejącymi testami klinicznymi.
Zaglądanie w przerwy i sygnały epigenetyczne
Powyżej liczenia powtórzeń, metoda bada drobną strukturę regionu powtórzeń i jego chemiczne oznakowanie. U niektórych pacjentów występują przerwy — małe fragmenty o innej sekwencji w obrębie powtórzenia — które mogą wiązać się z łagodniejszym przebiegiem choroby. Zespół stwierdził, że wiele pozornych przerw było w rzeczywistości artefaktami przetwarzania surowego sygnału i pokazał, że zastosowanie większych okien analitycznych podczas dekodowania znacząco zmniejsza liczbę fałszywych pozytywów. RepeatLab wykorzystuje też specjalny tryb odczytu metylacji DNA — chemicznego znacznika, który może wyłączać lub włączać geny. W rejonie genu DM1 autorzy zmapowali wzory metylacji na poziomie pojedynczych alleli i zidentyfikowali kilka kluczowych stref, gdzie silniejsza metylacja koreluje z dłuższymi ekspansjami powtórzeń, wspierając ideę, że te chemiczne markery wpływają na przebieg choroby.
Szybszy, bogatszy test dla trudnych do zmierzenia powtórzeń
W sumie praca pokazuje, że wzbogacanie kierowane przez CRISPR, sekwencjonowanie długich odczytów nanopore i przyjazne dla użytkownika oprogramowanie potrafią dostarczyć szczegółowy profil chorobotwórczych powtórzeń DNA w mniej niż 24 godziny. Dla DM1 i pokrewnych schorzeń podejście to mierzy nie tylko długość powtórzeń, lecz także wychwytuje subtelne zmiany sekwencyjne i wzory epigenetyczne, które w dużej mierze umykają obecnym testom szpitalnym. Choć potrzebna jest dalsza walidacja i dostosowanie do nowszych chemii sekwencjonowania, zintegrowana platforma wskazuje kierunek dla przyszłych badań genetycznych, które będą szybsze, bardziej informatywne i łatwiejsze do wdrożenia w rutynowej opiece.
Cytowanie: Han, Y., Jang, JH. & Chang, H. Targeted long-read sequencing for high-resolution repeat profiling in myotonic dystrophy type 1. Exp Mol Med 58, 1203–1215 (2026). https://doi.org/10.1038/s12276-026-01683-6
Słowa kluczowe: dystrofia miotoniczna, ekspansja powtórzeń tandemowych, sekwencjonowanie nanopore, wzbogacanie CRISPR Cas9, metylacja DNA