Clear Sky Science · fr
Séquençage ciblé en longues lectures pour un profilage haute résolution des répétitions dans la myotonie dystrophique de type 1
Pourquoi c’est important pour les familles et les médecins
Certaines maladies héréditaires sont causées par des segments « qui bégaient » dans notre ADN — de courtes séquences répétées des centaines voire des milliers de fois. Lorsque ces répétitions deviennent trop longues, elles peuvent entraîner des affections graves comme la myotonie dystrophique, une maladie entraînant une fonte musculaire. Pourtant, mesurer précisément la longueur de ces répétitions et leur marquage chimique est étonnamment difficile avec les tests hospitaliers actuels. Cet article présente une méthode simplifiée pour lire ces régions d’ADN difficiles en détail en environ une journée, offrant potentiellement aux médecins des réponses plus nettes et aux patients des conseils plus précis.
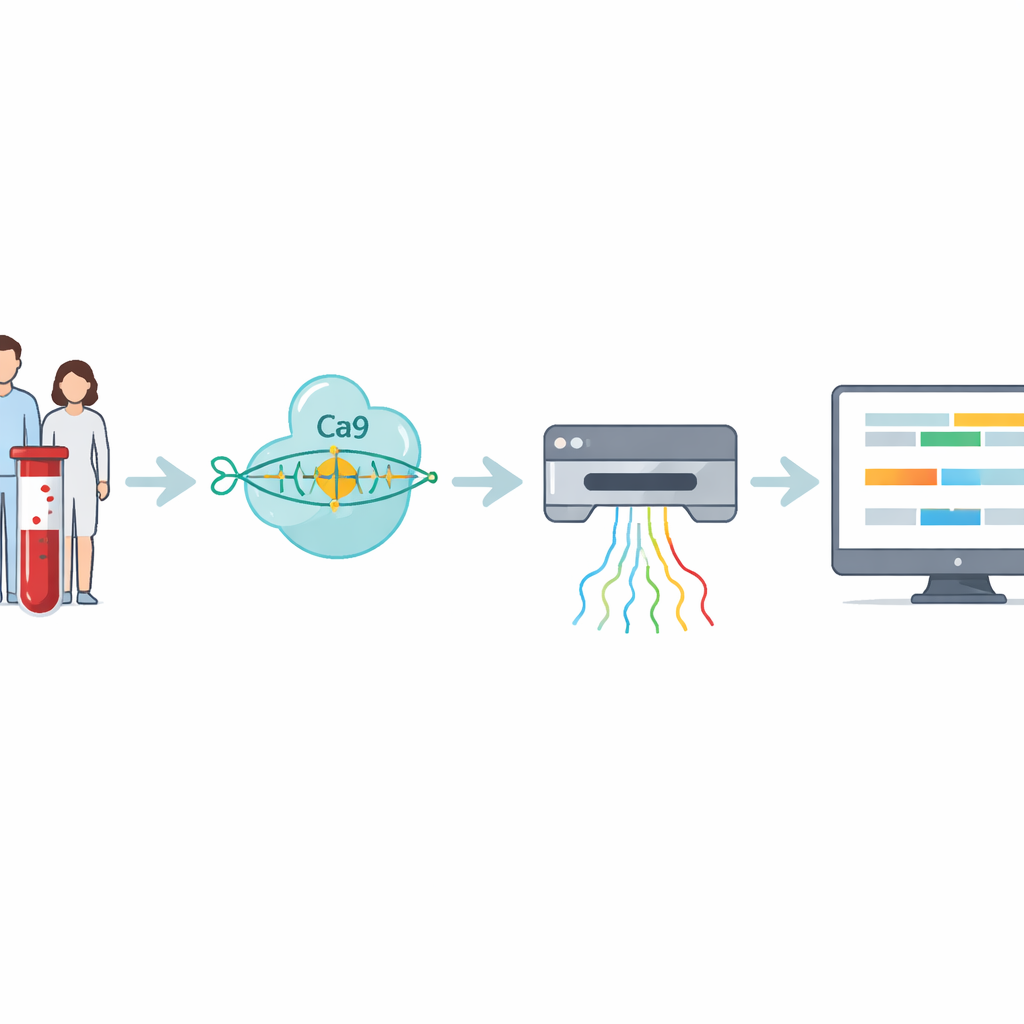
Un examen approfondi d’une maladie musculaire complexe
La myotonie dystrophique de type 1 (DM1) est causée par un allongement excessif d’un triplet de bases dans un gène important pour le muscle et d’autres tissus. Les personnes ayant davantage de répétitions présentent en général une forme plus sévère, de sorte que connaître la longueur exacte de la répétition est essentiel pour le diagnostic et le pronostic. Les outils standards des laboratoires cliniques — comme certains tests PCR spécialisés et les Southern blots — fonctionnent bien lorsque la répétition est modérée, mais ils échouent souvent ou deviennent très imprécis dès que l’expansion dépasse quelques centaines de copies. Cela crée une zone d’incertitude où les familles savent qu’une mutation est présente mais n’ont pas une image claire de son étendue ni de son évolution potentielle.
Combiner des ciseaux moléculaires et la lecture longue de l’ADN
Les auteurs ont développé un protocole en quatre étapes qui cible directement ces répétitions difficiles à lire. D’abord, l’ADN est extrait du sang ou d’échantillons cellulaires. Ensuite, l’équipe utilise CRISPR–Cas9 — des ciseaux moléculaires guidés vers une adresse spécifique du génome — pour couper autour de la région contenant la répétition. Comme seuls ces fragments coupés sont préparés pour le séquençage, la méthode enrichit fortement la région d’intérêt tout en évitant l’amplification par PCR, qui a tendance à manquer les très grandes expansions. L’ADN préparé est ensuite introduit dans un séquenceur nanopore, un appareil portatif qui fait passer les brins d’ADN à travers de minuscules pores et détecte leur composition en temps réel via des signaux électriques.
Un logiciel intelligent pour mesurer longueur et marques chimiques
Les signaux bruts du séquenceur doivent être traduits en code ADN puis en mesures exploitables. Ici, les auteurs présentent RepeatLab, une chaîne d’analyse automatisée conçue pour être utilisée même sur un simple notebook cloud. RepeatLab effectue d’abord un passage rapide pour repérer les lectures couvrant le gène ciblé, puis retraitement de ces lectures avec un réglage plus exigeant et de haute précision adapté aux longues régions répétitives. Il utilise une stratégie statistique modifiée pour regrouper les mesures mono-molécule en deux classes — en général la copie normale et la copie étendue du gène chez un individu — obtenant des estimations de longueur fiables avec aussi peu qu’une dizaine de lectures. Le même cadre peut analyser de nombreux échantillons ou plusieurs gènes en une seule exécution, en maintenant des coûts comparables aux tests cliniques existants.
Regarder les interruptions et les signaux épigénétiques
Au-delà du simple comptage des répétitions, la méthode examine la structure fine de la région répétée et sa décoration chimique. Certains patients portent des interruptions — de petits segments de séquence différents à l’intérieur de la répétition — qui peuvent être associés à une maladie plus bénigne. L’équipe a constaté que de nombreuses interruptions apparentes étaient en réalité des artefacts du traitement du signal brut, et a montré que l’utilisation de fenêtres d’analyse plus larges lors du décodage réduisait fortement ces faux positifs. RepeatLab propose aussi un mode spécialisé pour lire la méthylation de l’ADN, une marque chimique qui peut activer ou réprimer des gènes. Autour du gène DM1, les auteurs ont cartographié les schémas de méthylation à résolution mono-allèle et identifié plusieurs zones clés où une méthylation accrue coïncide avec des expansions de répétitions plus longues, soutenant l’idée que ces marques chimiques participent à la façon dont la maladie se manifeste.
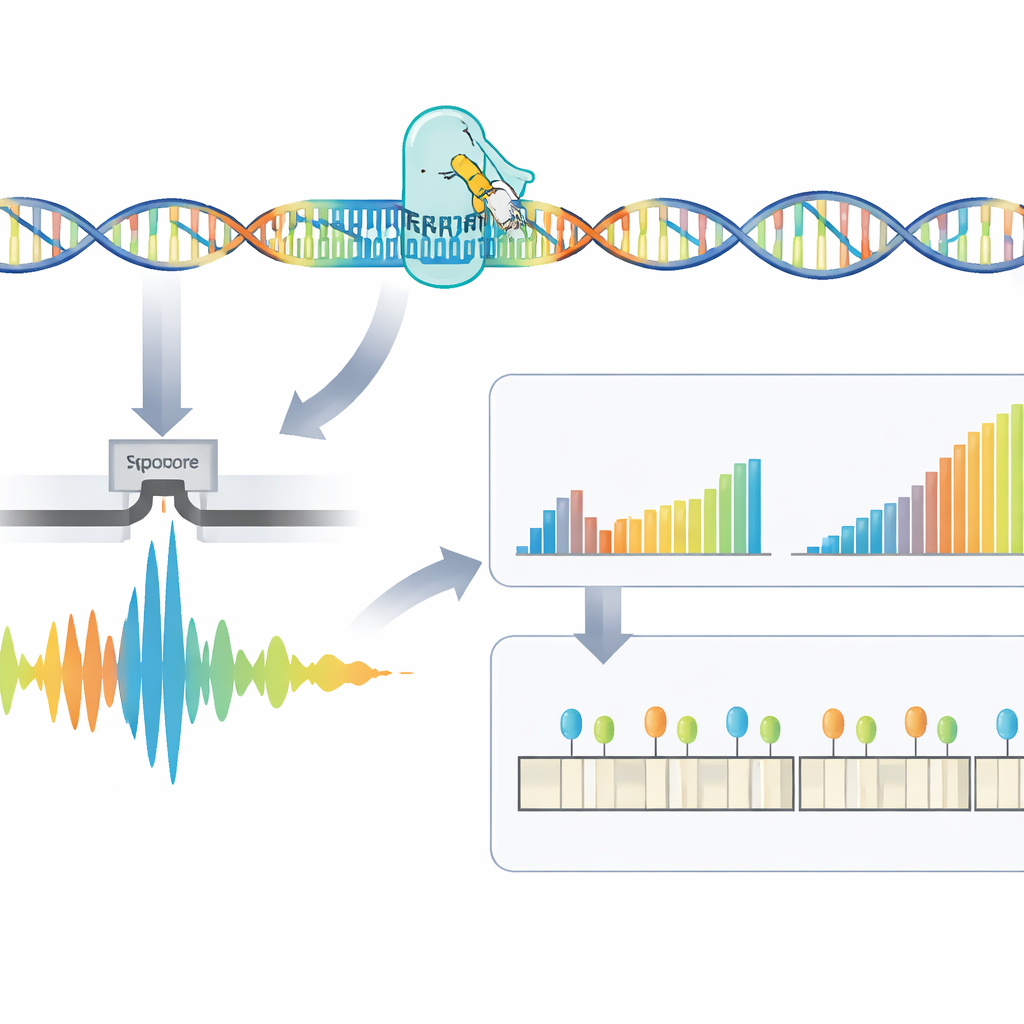
Un test plus rapide et plus riche pour les répétitions difficiles à mesurer
Pris ensemble, ces travaux montrent que l’enrichissement guidé par CRISPR, le séquençage long par nanopore et un logiciel convivial peuvent fournir un profil détaillé des répétitions d’ADN responsables de maladies en moins de 24 heures. Pour la DM1 et les affections apparentées, l’approche non seulement mesure la longueur des répétitions, mais capture aussi des changements de séquence subtils et des motifs épigénétiques que les tests hospitaliers actuels ne détectent pour la plupart pas. Bien que des validations supplémentaires et des adaptations aux nouvelles chimies de séquençage demeurent nécessaires, cette plateforme intégrée ouvre la voie à des tests génétiques futurs plus rapides, plus informatifs et plus faciles à déployer en routine clinique.
Citation: Han, Y., Jang, JH. & Chang, H. Targeted long-read sequencing for high-resolution repeat profiling in myotonic dystrophy type 1. Exp Mol Med 58, 1203–1215 (2026). https://doi.org/10.1038/s12276-026-01683-6
Mots-clés: myotonie dystrophique, expansion de répétitions en tandem, séquençage nanopore, enrichissement CRISPR Cas9, méthylation de l’ADN