Clear Sky Science · pt
Sequenciamento direcionado de leitura longa para perfil de alta resolução de repetições na distrofia miotônica tipo 1
Por que isso importa para famílias e médicos
Algumas doenças hereditárias são causadas por trechos “gaguejantes” no nosso DNA—sequências curtas repetidas centenas ou até milhares de vezes. Quando essas repetições crescem demais, podem provocar condições graves como a distrofia miotônica, uma doença que leva à degeneração muscular. Ainda assim, medir com precisão o tamanho dessas repetições e como elas estão quimicamente marcadas é surpreendentemente difícil com os testes hospitalares atuais. Este artigo apresenta um modo simplificado de ler essas regiões difíceis do DNA em detalhe em cerca de um dia, potencialmente oferecendo respostas mais claras aos médicos e orientação melhor aos pacientes.
Um olhar mais atento sobre uma doença muscular complexa
A distrofia miotônica tipo 1 (DM1) é causada por uma repetição excessiva de três letras do DNA em um gene importante para músculos e outros tecidos. Pessoas com mais repetições tendem a ter doença mais grave, então conhecer o comprimento exato da repetição é vital para diagnóstico e prognóstico. Ferramentas padrão em laboratórios clínicos—como testes PCR específicos e Southern blot—funcionam bem quando a repetição tem tamanho moderado, mas frequentemente falham ou tornam-se muito imprecisas quando as expansões excedem algumas centenas de cópias. Isso deixa uma zona cinzenta em que as famílias podem saber que existe uma mutação, mas não têm um quadro claro de quão extensa ela é ou como pode evoluir ao longo do tempo.
Combinando tesouras moleculares e leitura longa de DNA
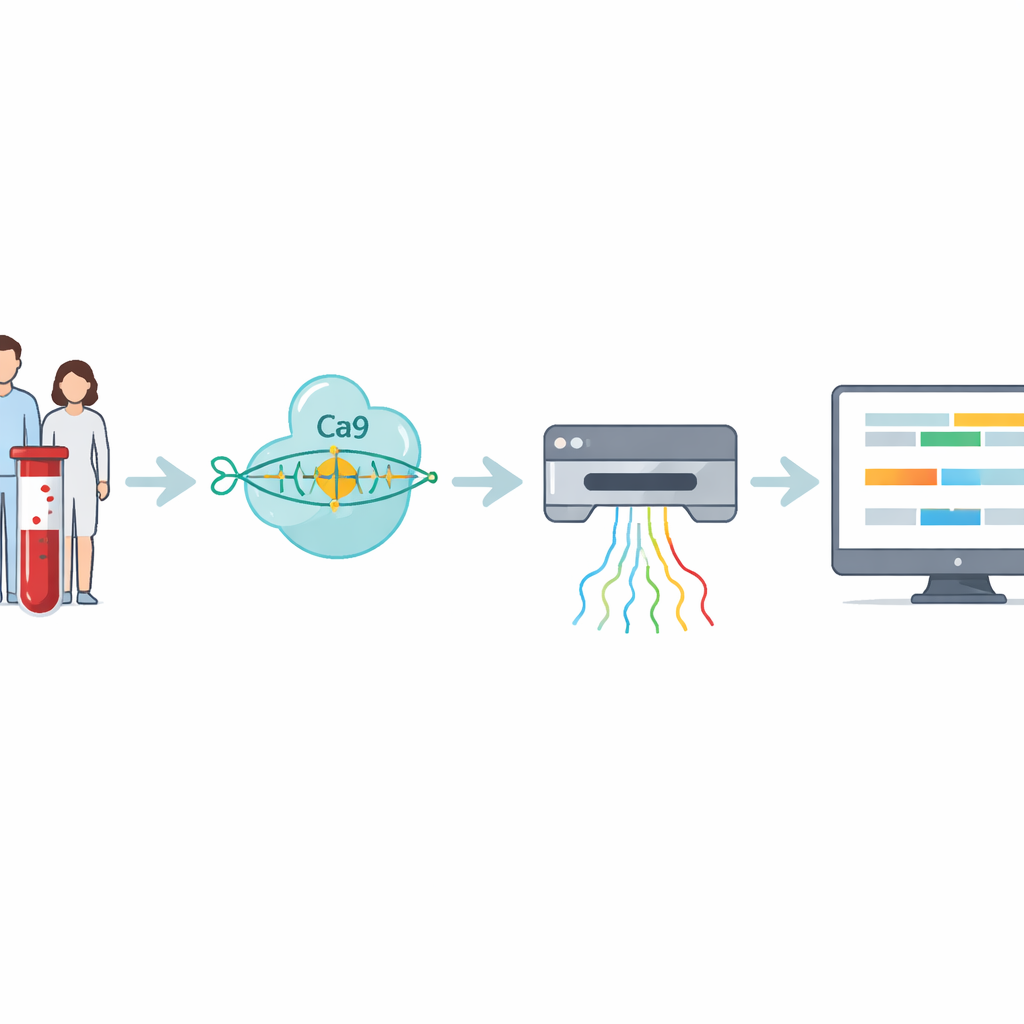
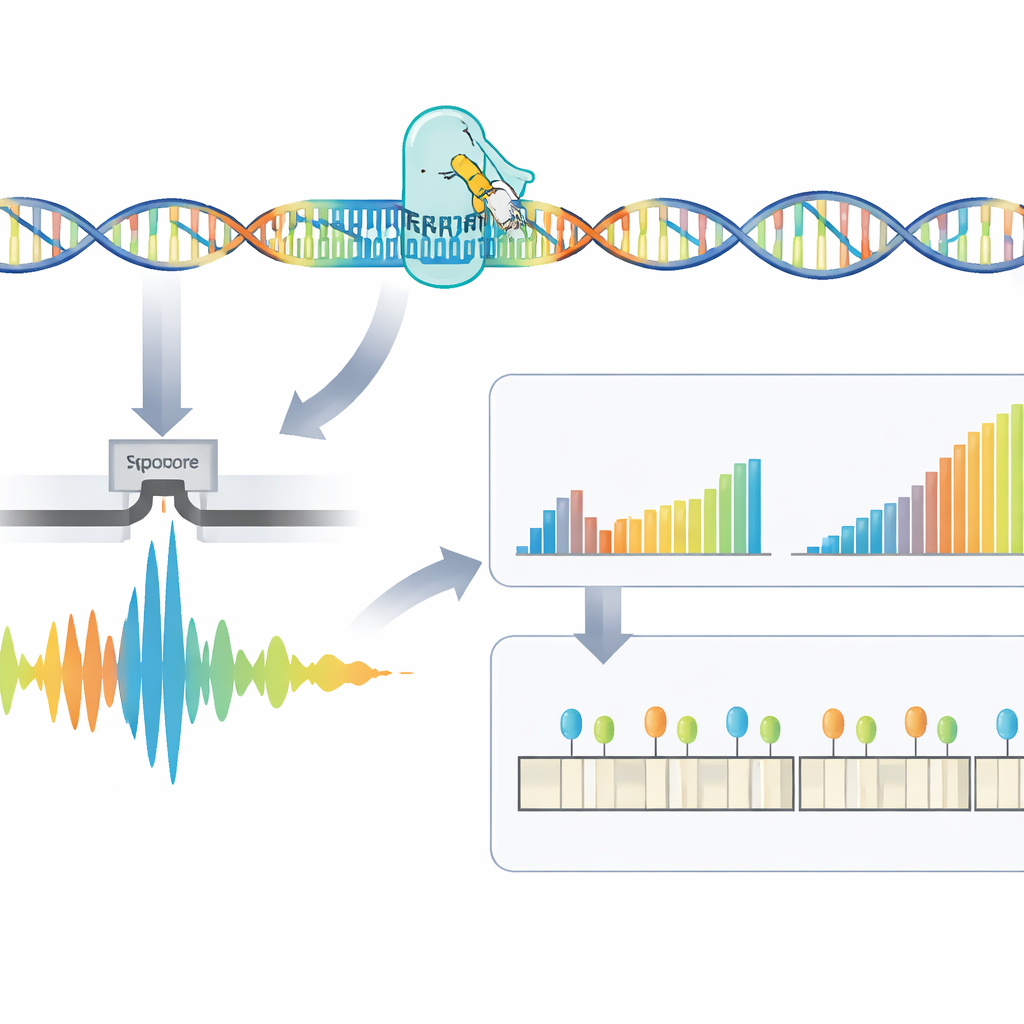
Os pesquisadores construíram um fluxo de trabalho em quatro etapas que ataca diretamente essas repetições difíceis de ler. Primeiro, o DNA é extraído de amostras de sangue ou de células. Em seguida, a equipe usa CRISPR–Cas9—tesouras moleculares guiadas a um endereço específico no genoma—para cortar em torno do trecho de DNA que contém a repetição. Como apenas esses fragmentos cortados são preparados para o sequenciamento, o método enriquece fortemente a região de interesse enquanto evita a cópia do DNA por PCR, que tende a perder expansões muito grandes. O DNA preparado é então submetido a um sequenciador por nanoporo, um dispositivo portátil que passa filamentos de DNA por poros minúsculos e detecta sua composição em tempo real por sinais elétricos.
Software inteligente para ler comprimento e marcas químicas
Os sinais brutos do sequenciador precisam ser traduzidos em código de DNA e depois em medidas significativas. Aqui os autores introduzem o RepeatLab, um pipeline de análise automatizado projetado para ser usado até em um notebook comum na nuvem. O RepeatLab faz primeiro uma varredura rápida para encontrar leituras que cobrem o gene alvo e depois reprocessa essas leituras com uma configuração mais exigente e de alta precisão, adaptada a trechos longos e repetitivos. Ele usa uma estratégia estatística modificada para agrupar medidas de moléculas únicas em dois grupos—normalmente as cópias normal e expandida do gene em cada pessoa—alcançando estimativas de comprimento confiáveis com tão poucas quanto cerca de uma dúzia de leituras. A mesma estrutura pode analisar muitas amostras ou vários genes em uma só execução, mantendo custos comparáveis aos exames clínicos existentes.
Investigando interrupções e sinais epigenéticos
Além de simplesmente contar repetições, o método examina a estrutura fina da região repetida e como ela está quimicamente decorada. Alguns pacientes têm interrupções—pequenos trechos de sequência diferente dentro da repetição—que podem estar ligados a formas mais brandas da doença. A equipe descobriu que muitas interrupções aparentes eram, na verdade, artefatos do processamento do sinal bruto, e mostrou que usar janelas de análise maiores durante a decodificação reduziu muito esses falsos positivos. O RepeatLab também usa um modo especializado para ler a metilação do DNA, uma marca química que pode ativar ou desativar genes. Ao redor do gene DM1, os autores mapearam padrões de metilação em resolução de alelo único e identificaram várias zonas-chave onde metilação mais intensa se correlaciona com expansões de repetição mais longas, apoiando a ideia de que essas marcas químicas ajudam a moldar como a doença se manifesta.
Um teste mais rápido e mais informativo para repetições difíceis de medir
Em conjunto, este trabalho mostra que o enriquecimento guiado por CRISPR, o sequenciamento por leitura longa por nanoporo e um software amigável podem fornecer um perfil detalhado de repetições de DNA causadoras de doença em menos de 24 horas. Para DM1 e condições relacionadas, a abordagem não apenas mede o comprimento das repetições, mas também captura mudanças sutis na sequência e padrões epigenéticos que os testes hospitalares atuais em grande parte não detectam. Embora sejam necessários mais validações e adaptações às químicas de sequenciamento mais recentes, essa plataforma integrada aponta para testes genéticos futuros que sejam mais rápidos, mais informativos e mais fáceis de implantar na prática clínica rotineira.
Citação: Han, Y., Jang, JH. & Chang, H. Targeted long-read sequencing for high-resolution repeat profiling in myotonic dystrophy type 1. Exp Mol Med 58, 1203–1215 (2026). https://doi.org/10.1038/s12276-026-01683-6
Palavras-chave: distrofia miotônica, expansão de repetição tandem, sequenciamento por nanoporo, enriquecimento CRISPR Cas9, metilação do DNA