Clear Sky Science · ru
Новый гомозиготный сплайсинговый вариант в FRA10AC1: дальнейшее уточнение фенотипа
Когда редактирование генов даёт сбой
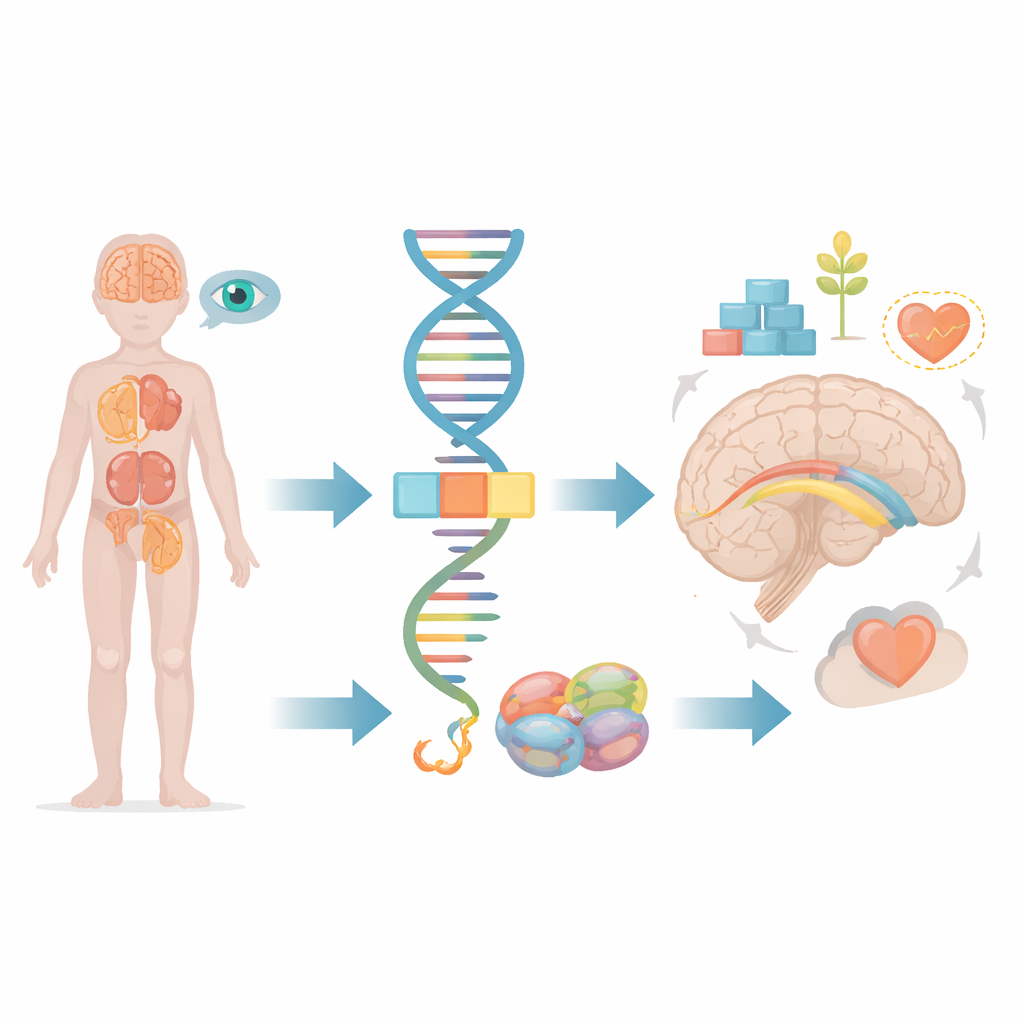
Большинство из нас воспринимают гены как неизменные чертежи, но на самом деле клетки постоянно вырезают и склеивают генетические сообщения, прежде чем превратить их в рабочие белки. В этой статье исследуется, что происходит, когда небольшая ошибка в этом процессе редактирования затрагивает ген FRA10AC1 и приводит к серьёзным нарушениями развития у детей. Проследив историю одного ребёнка и сравнив её с несколькими известными случаями в мире, исследователи показывают, как тонкий дефект в обработке генетической информации может отразиться на мозге, почках, глазах и других органах.
Маленькие редакторы клетки
Внутри каждой клетки длинные участки сырого генетического кода необходимо обрезать и соединить, прежде чем они смогут использоваться. Эту задачу выполняет молекулярный аппарат, известный как сплайсосома, который вырезает ненужные сегменты и сшивает полезные. Ген FRA10AC1 кодирует вспомогательный белок, который находится на периферии этого аппарата и тонко настраивает процесс вырезания и склейки. Предыдущие исследования показали, что дети, унаследовавшие дефектные копии FRA10AC1 от обоих родителей, могут развивать характерный набор признаков: задержку развития, необычные черты лица, плохой рост и изменения в плотном пучке нервных волокон, соединяющем две половины мозга — мозолистом теле. Однако к настоящему времени описано лишь десять таких пациентов во всём мире, и остаются вопросы о полном спектре симптомов и о том, как разные типы повреждений гена влияют на исход.
История одного ребёнка добавляет новые фрагменты
Авторы описывают мальчика египетского происхождения, родившегося у близкородственных родителей после протекшей без осложнений беременности. С раннего детства он демонстрировал выраженную задержку во всех областях развития: в восемь месяцев он не улыбался, не следил глазами, не реагировал на звуки и не контролировал голову. Врачи отмечали очень низкий мышечный тонус, нестабильные движения корпуса и быстрые неконтролируемые движения глаз — нистагм. Во внешности были характерные черты: вытянутое лицо, высокий лоб, складки кожи во внутренних уголках глаз, удлинённые глазные щели, уплощённый переносица и округлый кончик носа с длинной верхней губой. По мере роста он смог встать и произнести несколько простых слогов, но оставался тяжело отстающим в развитии и очень гиперактивным.
Скрытые изменения в мозге, глазах и почках
Сканирование мозга ребёнка выявило несколько структурных особенностей: недоразвитие соединяющего моста между полушариями, задержку формирования миелиновой оболочки вокруг нервных волокон и небольшую заполненную жидкостью кисту глубоко в области хвостатого ядра. Офтальмологическое обследование показало повреждение колбочек сетчатки, необходимых для острого центрального и цветового зрения, что объясняет его слабые зрительные реакции. УЗИ брюшной полости выявило ещё одно необычное нахождение: обе почки были сращены и располагались низко в области таза, а не на привычных местах. Эти поражения почек и глаз раньше встречались лишь изредка в описаниях случаев, связанных с FRA10AC1, поэтому команда тщательно проанализировала весь набор белок-кодирующих генов ребёнка в поисках других правдоподобных объяснений и не обнаружила таковых.
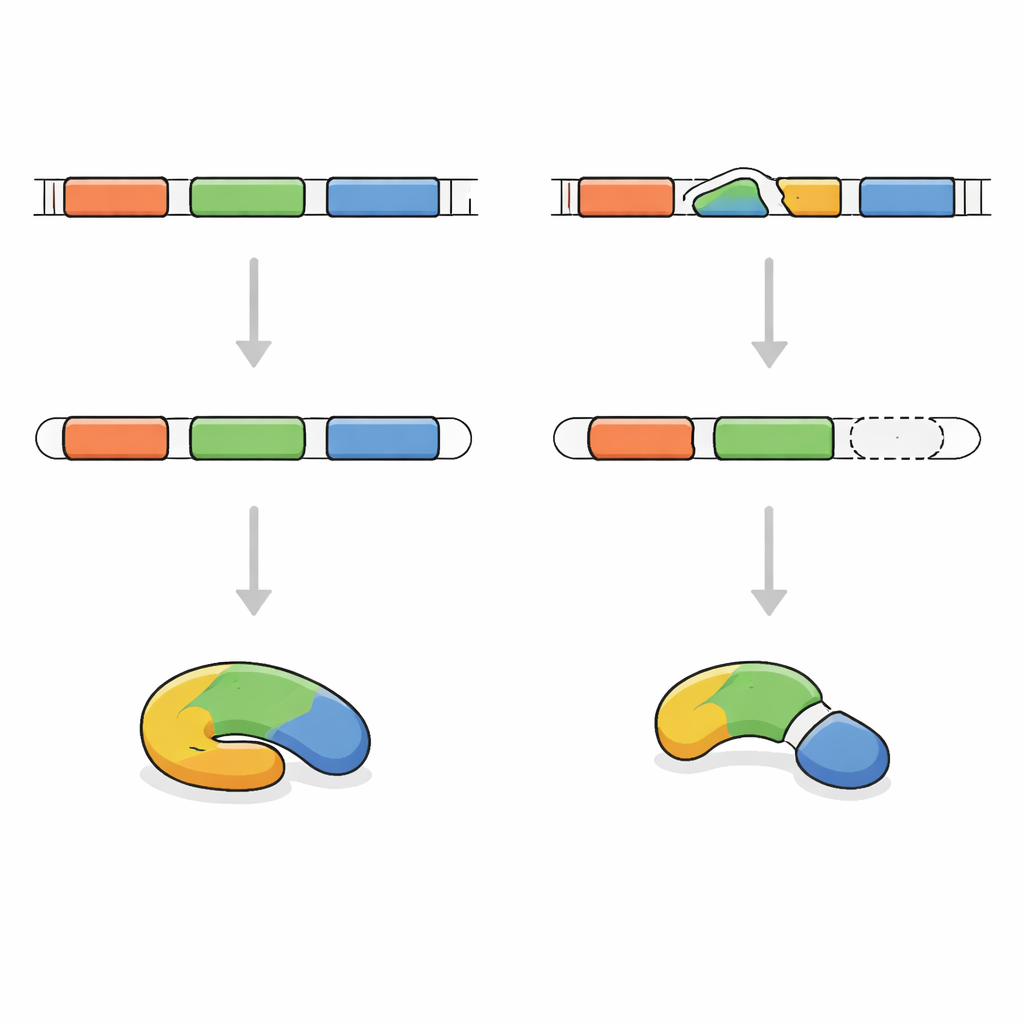
Отслеживание неверного генетического сообщения
Чтобы выяснить причину, исследователи секвенировали белок-кодирующие участки ДНК мальчика. Они обнаружили, что обе копии его гена FRA10AC1 несут одну и ту же небольшую замену в критической «точке соединения», используемой при редактировании сообщения. Родители, которые были здоровы, каждый несли по одной изменённой и одной нормальной копии. Затем команда изучила транскрипт FRA10AC1, полученный из его клеток крови. Вместо ожидаемого полного набора сегментов в сообщении один фрагмент полностью пропустился. Это вызвало сдвиг рамки считывания, введя ранний стоп-сигнал и приведя к укороченному, вероятно нефункциональному белку. Согласно международным рекомендациям по интерпретации генетических вариантов, это изменение классифицировано как однозначно вызывающее заболевание.
Что это значит для семей
Добавив случай этого мальчика к небольшому числу уже известных, исследование помогает прояснить картину расстройства, связанного с FRA10AC1. Дети с полным отсутствием функционирующего белка, как этот пациент, обычно имеют более тяжёлые когнитивные и моторные нарушения и с большей вероятностью сопровождаются врождёнными аномалиями других органов, таких как сердце, почки, глаза и кожа. Напротив, ранее описанные дети с более мягким, частично функционирующим вариантом FRA10AC1 имели менее выраженные нарушения обучения и меньше аномалий органов. Для семей и клиницистов эта работа подчёркивает, что дефект в одном гене может лежать в основе узнаваемого сочетания черт лица, задержек развития и изменений в мозге, и что при подозрении на диагноз важны тщательные обследования почек и глаз.
Цитирование: Abdel-Hamid, M.S., Abdel-Salam, G.M.H. A novel homozygous splicing variant in FRA10AC1: further delineation of the phenotype. J Hum Genet 71, 363–367 (2026). https://doi.org/10.1038/s10038-025-01447-6
Ключевые слова: нарушение нейроразвития, сплайсосома, ген FRA10AC1, генетические варианты, мозолистое тело