Clear Sky Science · pt
Uma nova variante de splicing homozigótica em FRA10AC1: maior delimitação do fenótipo
Quando a edição genética dá errado
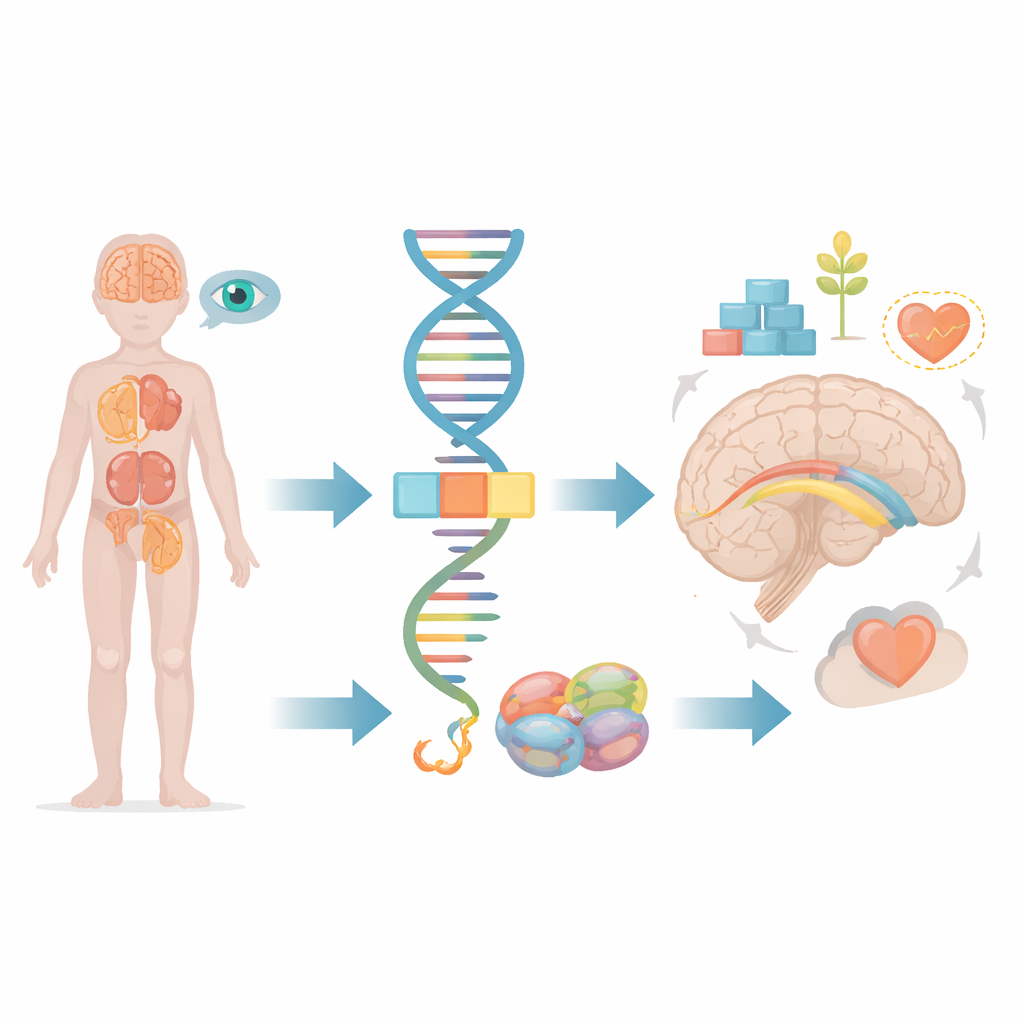
A maioria de nós pensa nos genes como plantas estáticas, mas, na realidade, nossas células estão constantemente cortando e colando mensagens genéticas antes de transformá-las em proteínas funcionais. Este artigo explora o que ocorre quando uma pequena falha nesse processo de edição afeta um gene chamado FRA10AC1, levando a problemas de desenvolvimento graves em crianças. Ao acompanhar um paciente infantil e compará‑lo com os poucos casos conhecidos no mundo, os pesquisadores mostram como uma falha sutil no processamento da informação genética pode se espalhar até o cérebro, rins, olhos e outros órgãos.
Os pequenos editores da célula
Dentro de cada célula, longos trechos de código genético bruto precisam ser aparados e unidos antes de poderem ser utilizados. Essa tarefa é realizada por uma máquina molecular conhecida como espliceossomo, que remove segmentos não usados e costura os trechos úteis. O gene FRA10AC1 produz uma proteína auxiliar que se assenta na borda desta máquina e ajusta finamente como os cortes e junções ocorrem. Estudos anteriores descobriram que crianças que herdam cópias defeituosas do FRA10AC1 de ambos os pais podem desenvolver um padrão distinto de atraso no desenvolvimento, feições faciais incomuns, crescimento deficiente e alterações no feixe espesso de fibras nervosas que conecta as duas metades do cérebro, chamado corpo caloso. No entanto, apenas dez desses pacientes haviam sido descritos em todo o mundo, deixando muitas perguntas sobre o espectro completo de sintomas e como diferentes tipos de falhas no gene moldam o desfecho.
A história de uma criança adiciona novas peças
Os autores descrevem um menino egípcio nascido de pais aparentados após uma gravidez sem complicações. Desde a primeira infância ele apresentou atraso marcado em todas as áreas do desenvolvimento: não sorria, não seguia com os olhos, não reagia a sons nem desenvolveu controle de cabeça aos oito meses de idade. Os médicos notaram tônus muscular muito baixo, movimentos do tronco instáveis e movimentos oculares rápidos e descontrolados chamados nistagmo. Sua aparência facial incluía rosto alongado, testa alta, pregas de pele nos cantos internos dos olhos, aberturas oculares longas, dorso nasal achatado e ponta nasal arredondada com lábio superior alongado. À medida que cresceu, ele acabou conseguindo ficar em pé e pronunciar algumas sílabas simples, mas permaneceu com atraso severo e muito hiperativo.
Mudanças ocultas no cérebro, olhos e rins
Exames de imagem do cérebro da criança revelaram várias diferenças estruturais: a ponte de conexão entre as duas metades do cérebro estava subdesenvolvida, o material isolante ao redor das fibras nervosas apresentava atraso na formação e havia um pequeno cisto preenchido por líquido em profundidade na região chamada núcleo caudado. Exames oculares mostraram dano às células cones da retina, necessárias para visão central nítida e percepção de cores, o que explica suas respostas visuais pobres. A imagem do abdome revelou outra descoberta incomum: ambos os rins estavam fundidos e localizados baixos na pelve, em vez das posições habituais. Esses problemas renais e oculares haviam sido observados apenas raramente em casos anteriores relacionados ao FRA10AC1, de modo que a equipe examinou cuidadosamente todo o conjunto de genes codificadores de proteínas da criança em busca de quaisquer outras explicações plausíveis e não encontrou nenhuma.
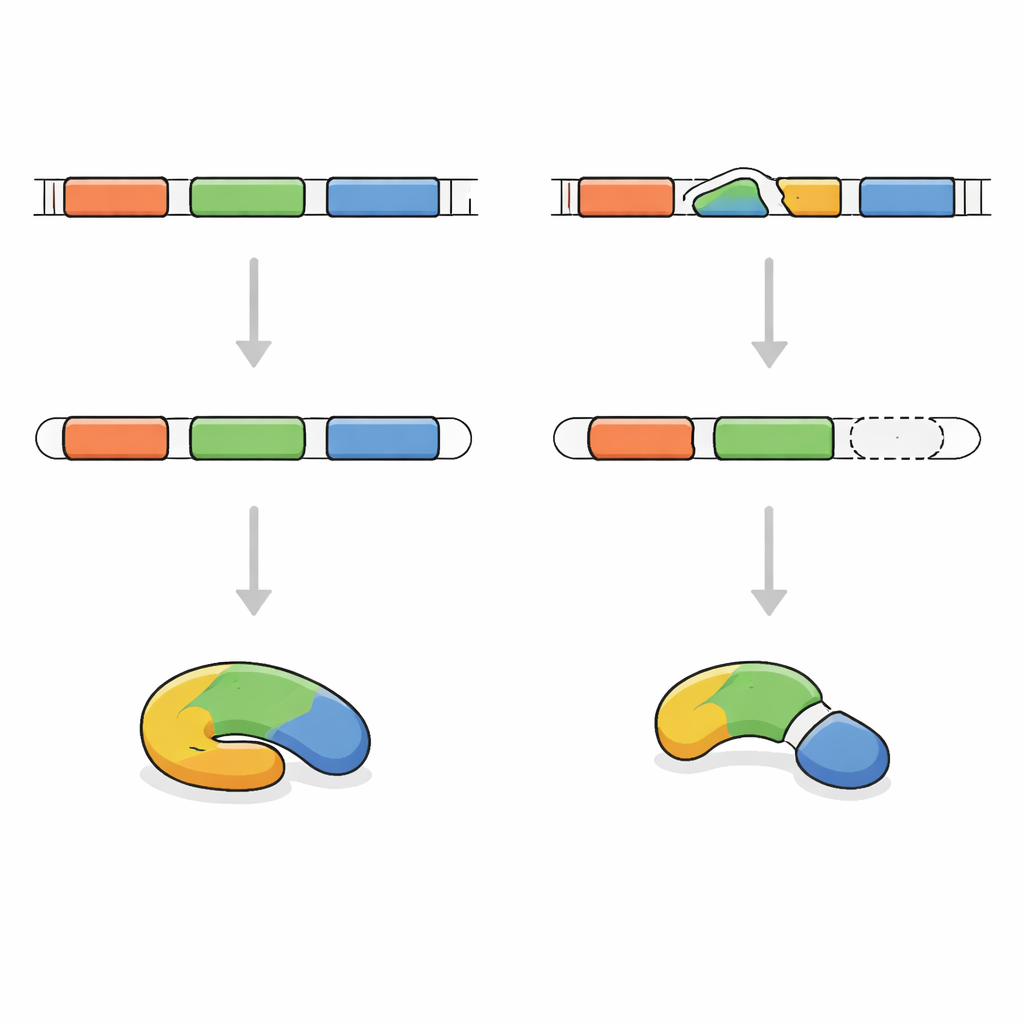
Rastreando a mensagem genética defeituosa
Para identificar a causa, os pesquisadores sequenciaram as porções codificadoras de proteínas do DNA do menino. Eles descobriram que ambas as cópias do gene FRA10AC1 carregavam a mesma pequena alteração em um ponto crítico de "junção" usado durante a edição da mensagem. Seus pais, saudáveis, eram portadores, cada um com uma cópia alterada e uma normal. A equipe então examinou a mensagem de FRA10AC1 produzida a partir das células sanguíneas dele. Em vez de incluir todos os segmentos esperados, a mensagem havia pulado uma parte inteira. Isso fez com que as instruções ficassem deslocadas, introduzindo um sinal de parada precoce e levando a uma proteína encurtada, provavelmente inútil. Com base em diretrizes internacionais para avaliação de variantes genéticas, essa alteração foi classificada como claramente causadora de doença.
O que isso significa para as famílias
Ao adicionar o caso desse menino ao pequeno número já conhecido, o estudo ajuda a esclarecer o quadro do distúrbio relacionado ao FRA10AC1. Crianças com perda completa da proteína, como este paciente, tendem a apresentar dificuldades intelectuais e motoras mais severas e têm maior probabilidade de apresentar defeitos congênitos adicionais que afetam órgãos como coração, rins, olhos e pele. Em contraste, crianças relatadas anteriormente com uma versão mais branda e parcialmente funcional do FRA10AC1 apresentaram problemas de aprendizagem menos graves e menos anomalias orgânicas. Para famílias e clínicos, este trabalho enfatiza que uma falha nesse gene único pode estar na base de uma combinação reconhecível de feições faciais, atrasos no desenvolvimento e alterações cerebrais, e que verificações cuidadosas dos rins e dos olhos são importantes quando o diagnóstico é suspeitado.
Citação: Abdel-Hamid, M.S., Abdel-Salam, G.M.H. A novel homozygous splicing variant in FRA10AC1: further delineation of the phenotype. J Hum Genet 71, 363–367 (2026). https://doi.org/10.1038/s10038-025-01447-6
Palavras-chave: transtorno do neurodesenvolvimento, espliceossomo, gene FRA10AC1, variantes genéticas, corpo caloso