Clear Sky Science · fr
Un nouveau variant d’épissage homozygote dans FRA10AC1 : précision supplémentaire du phénotype
Quand l’édition génétique tourne mal
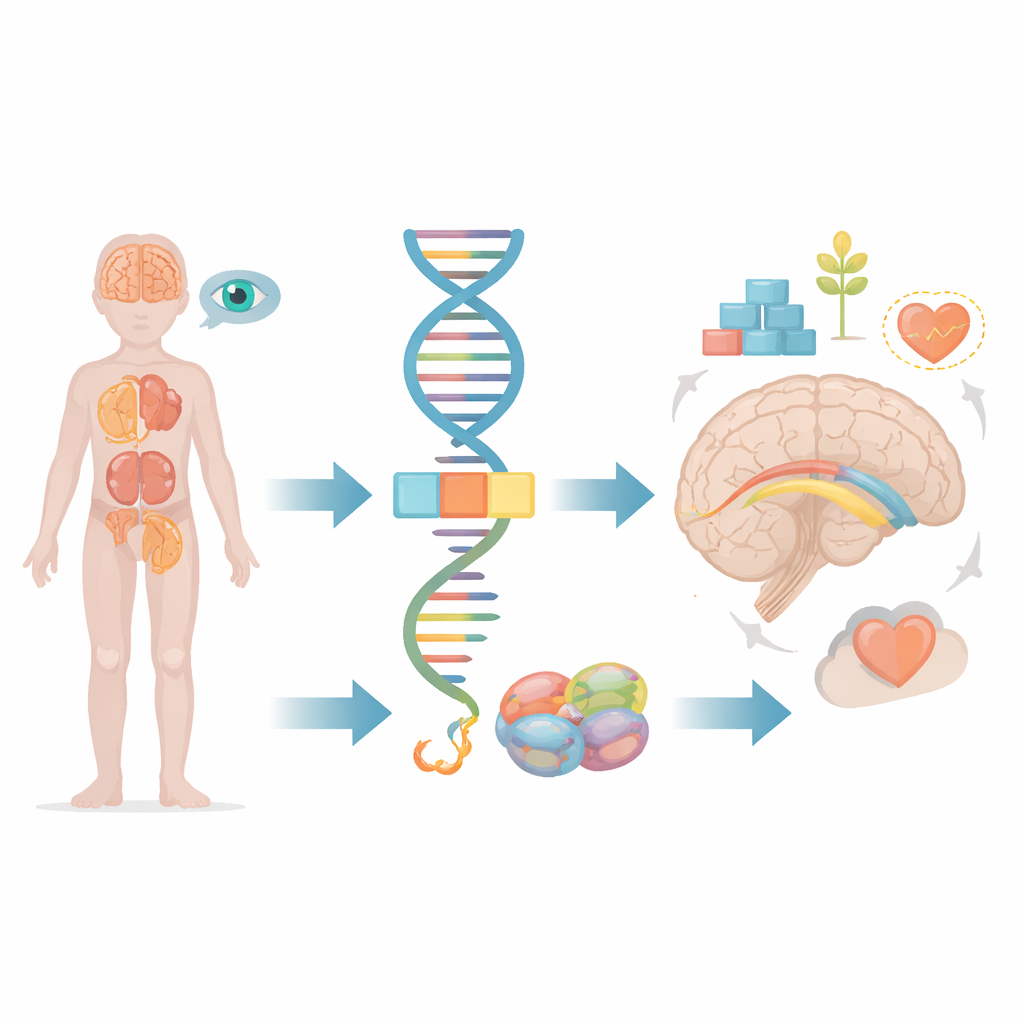
La plupart d’entre nous considèrent les gènes comme des plans fixes, mais en réalité nos cellules coupent et recollent continuellement les messages génétiques avant de les transformer en protéines actives. Cet article examine ce qui se passe lorsqu’une toute petite défaillance dans ce processus d’édition affecte un gène appelé FRA10AC1, entraînant des troubles développementaux graves chez des enfants. En suivant un jeune patient et en le comparant aux rares cas connus dans le monde, les chercheurs montrent comment un dysfonctionnement subtil du traitement de l’information génétique peut se répercuter sur le cerveau, les reins, les yeux et d’autres organes.
Les petits éditeurs de la cellule
À l’intérieur de chaque cellule, de longues séquences de code génétique brut doivent être retaillées et rassemblées avant de pouvoir être utilisées. Cette tâche est assurée par une machinerie moléculaire appelée spliceosome, qui élimine les segments inutiles et relie les segments utiles. Le gène FRA10AC1 produit une protéine auxiliaire qui se place en bordure de cette machinerie et ajuste finement la façon dont les découpes et les jonctions s’effectuent. Des études antérieures ont montré que des enfants héritant de copies défectueuses de FRA10AC1 de leurs deux parents peuvent développer un tableau caractéristique : retard global du développement, traits faciaux atypiques, croissance insuffisante et anomalies du faisceau épais de fibres nerveuses reliant les deux hémisphères du cerveau, appelé corps calleux. Cependant, seuls dix patients de ce type avaient été décrits dans le monde, laissant de nombreuses questions sur l’étendue complète des symptômes et sur la façon dont différents types de défauts dans le gène influencent le pronostic.
Le récit d’un enfant apporte de nouveaux éléments
Les auteurs décrivent un garçon égyptien né de parents consanguins après une grossesse sans complication. Dès la petite enfance, il montrait un retard marqué dans tous les domaines du développement : il ne souriait pas, ne suivait pas des yeux, ne répondait pas aux sons et n’avait pas acquis le contrôle de sa tête à huit mois. Les médecins ont noté un tonus musculaire très faible, des mouvements du tronc instables et des mouvements oculaires rapides et incontrôlés appelés nystagmus. Son visage présentait un allongement, un front haut, des plis de peau au coin interne des yeux, des ouvertures palpébrales allongées, un pont nasal aplati et une pointe du nez arrondie avec une lèvre supérieure longue. En grandissant, il a fini par se tenir debout et prononcer quelques syllabes simples, mais il est demeuré sévèrement retardé et très hyperactif.
Modifications cachées du cerveau, des yeux et des reins
Les examens du cerveau de l’enfant ont révélé plusieurs différences structurelles : le pont de connexion entre les deux hémisphères était sous-développé, la formation de la gaine isolante autour des fibres nerveuses était retardée, et une petite kyste rempli de liquide se trouvait en profondeur dans une région appelée noyau caudé. Les examens oculaires ont montré des lésions des cônes de la rétine, nécessaires à la vision centrale fine et à la perception des couleurs, ce qui explique ses réponses visuelles faibles. L’imagerie de l’abdomen a mis en évidence une autre anomalie peu commune : les deux reins étaient fusionnés et situés bas dans le bassin au lieu de leur position habituelle. Ces anomalies rénales et oculaires n’avaient été observées que rarement dans les cas FRA10AC1 précédents, si bien que l’équipe a scruté l’ensemble des gènes codant des protéines de l’enfant pour rechercher toute autre explication plausible et n’en a trouvé aucune.
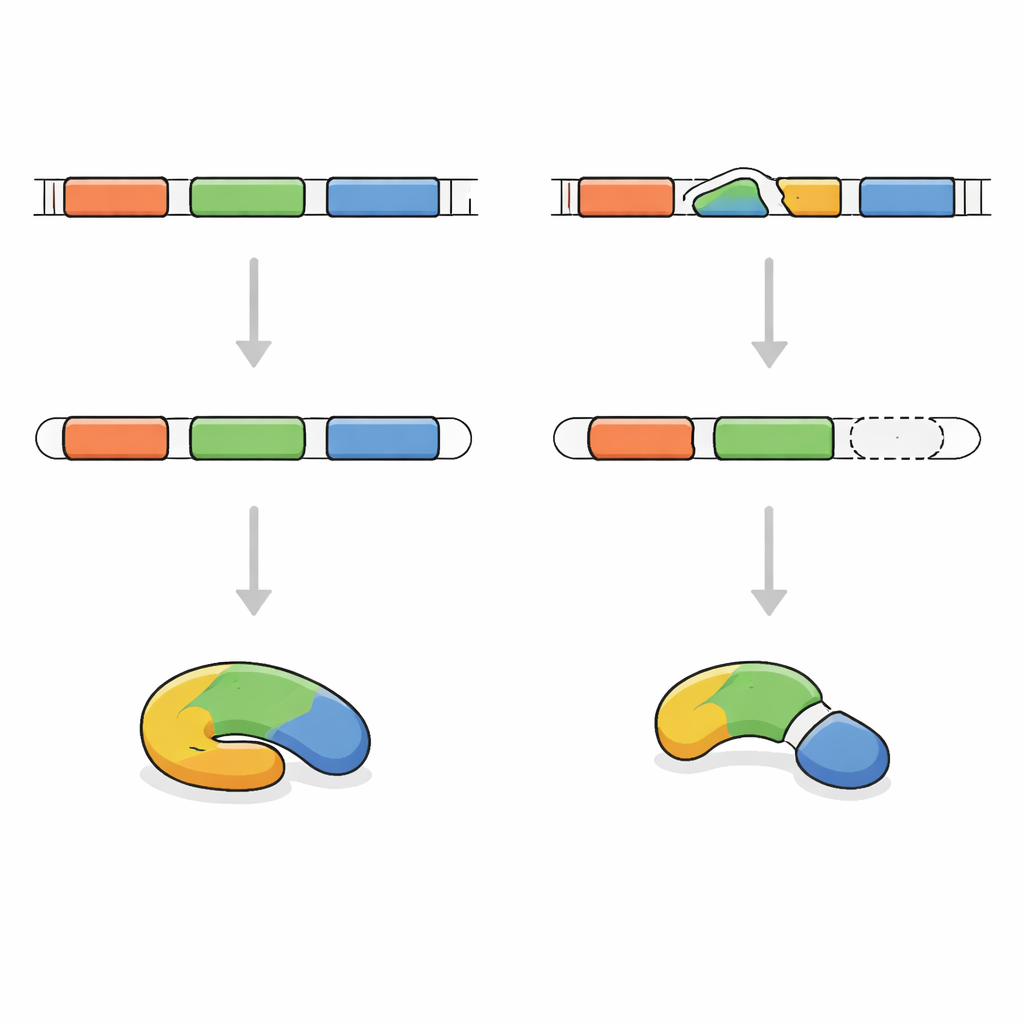
Suivre le message génétique défectueux
Pour identifier la cause, les chercheurs ont séquencé les portions codantes du génome du garçon. Ils ont découvert que ses deux copies du gène FRA10AC1 portaient la même petite modification à un point de jonction critique utilisé lors de l’édition du message. Ses parents, en bonne santé, étaient chacun porteurs d’une copie modifiée et d’une copie normale. L’équipe a ensuite examiné le message issu de FRA10AC1 produit par ses cellules sanguines. Au lieu d’inclure tous les segments attendus, le message avait sauté complètement un fragment. Cela a provoqué un décalage du cadre de lecture, introduisant un signal d’arrêt précoce et entraînant une protéine raccourcie probablement non fonctionnelle. D’après les lignes directrices internationales pour l’évaluation des variants génétiques, cette modification a été classée comme clairement pathogène.
Ce que cela signifie pour les familles
En ajoutant le cas de ce garçon au petit nombre déjà connu, l’étude contribue à préciser le tableau du trouble lié à FRA10AC1. Les enfants présentant une perte complète de la protéine, comme ce patient, tendent à présenter des déficits intellectuels et moteurs plus sévères et sont plus susceptibles d’avoir des malformations associées touchant des organes tels que le cœur, les reins, les yeux et la peau. En revanche, les enfants décrits antérieurement avec une version atténuée et partiellement fonctionnelle de FRA10AC1 avaient des troubles d’apprentissage moins sévères et moins d’anomalies d’organes. Pour les familles et les cliniciens, ce travail souligne qu’un défaut d’un seul gène peut être à l’origine d’une combinaison reconnaissable de traits faciaux, de retards de développement et d’anomalies cérébrales, et que des examens attentifs des reins et des yeux sont importants dès que le diagnostic est suspecté.
Citation: Abdel-Hamid, M.S., Abdel-Salam, G.M.H. A novel homozygous splicing variant in FRA10AC1: further delineation of the phenotype. J Hum Genet 71, 363–367 (2026). https://doi.org/10.1038/s10038-025-01447-6
Mots-clés: trouble du développement neurologique, spliceosome, gène FRA10AC1, variants génétiques, corps calleux