Clear Sky Science · de
Eine neue homozygote Spleißvariante in FRA10AC1: weitere Abgrenzung des Phänotyps
Wenn Gen-Editing schiefgeht
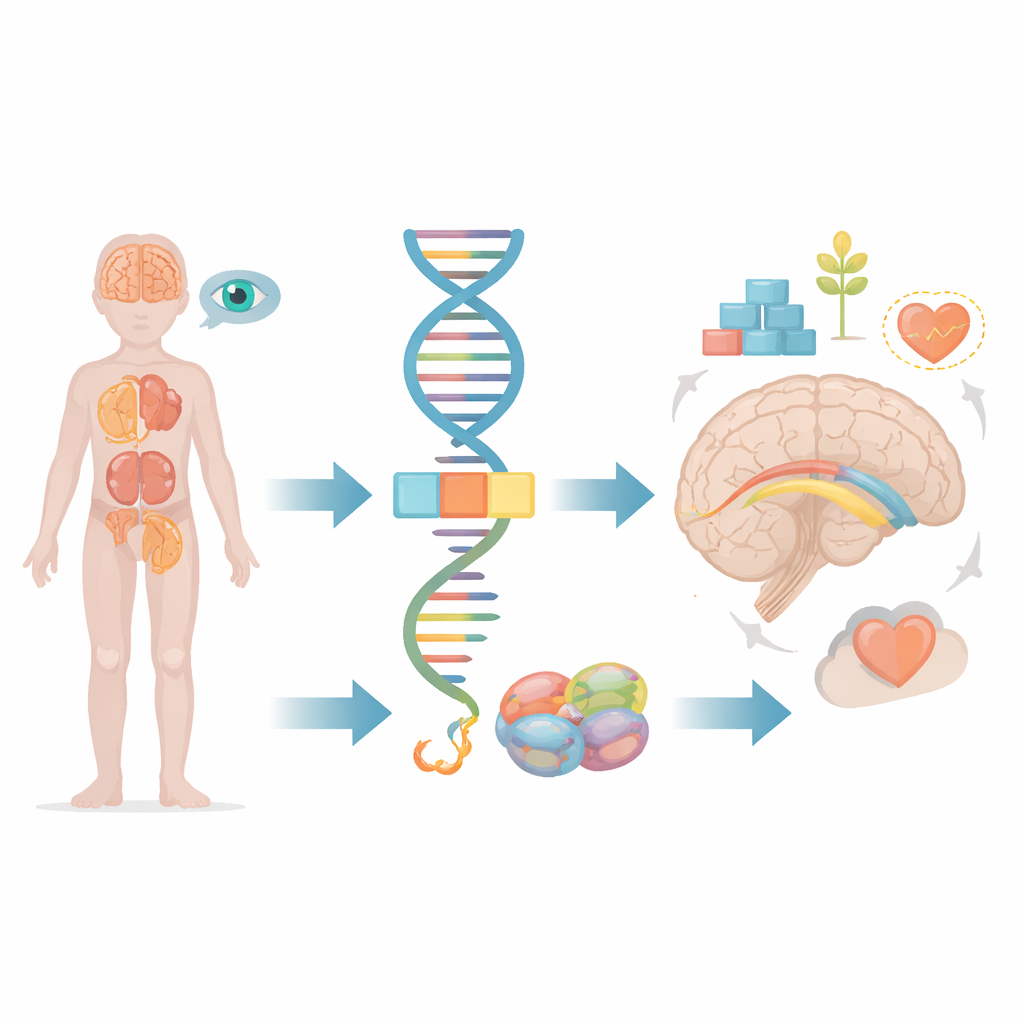
Die meisten von uns denken an Gene als statische Baupläne, doch in Wirklichkeit schneiden und kleben unsere Zellen ständig genetische Botschaften, bevor sie diese in funktionelle Proteine umsetzen. Dieser Artikel untersucht, was passiert, wenn ein winziger Fehler in diesem Editierprozess ein Gen namens FRA10AC1 betrifft und bei Kindern zu schweren Entwicklungsstörungen führt. Anhand der Beobachtung eines jungen Patienten und dem Vergleich mit wenigen weltweit bekannten Fällen zeigen die Forschenden, wie eine subtile Störung in der Informationsverarbeitung weitreichende Folgen für Gehirn, Nieren, Augen und andere Organe haben kann.
Die winzigen Editoren der Zelle
In jeder Zelle müssen lange Abschnitte ungefilterter genetischer Information zugeschnitten und zusammengefügt werden, bevor sie nutzbar sind. Diese Aufgabe übernimmt eine molekulare Maschine, das Spleißosom, das nicht benötigte Segmente herausschneidet und die nützlichen Stücke zusammennäht. Das FRA10AC1-Gen stellt ein Hilfsprotein her, das am Rand dieser Maschine sitzt und feinjustiert, wie Schneiden und Zusammenfügen ablaufen. Frühere Studien zeigten, dass Kinder, die von beiden Eltern fehlerhafte Kopien von FRA10AC1 erben, ein charakteristisches Muster aus verzögerter Entwicklung, auffälligen Gesichtsmerkmalen, Minderwuchs und Veränderungen im dicken Nervenfaserbündel, das die beiden Hirnhälften verbindet (Corpus callosum), entwickeln können. Bislang waren allerdings nur zehn solche Patienten weltweit beschrieben, sodass viele Fragen zum vollen Spektrum der Symptome und zur Auswirkung unterschiedlicher Genfehler offenblieben.
Eine Krankengeschichte ergänzt das Bild
Die Autorinnen und Autoren beschreiben einen ägyptischen Jungen, geboren von verwandten Eltern nach einer unkomplizierten Schwangerschaft. Schon im Säuglingsalter zeigte er ausgeprägte Verzögerungen in allen Entwicklungsbereichen: Mit acht Monaten lächelte er nicht, folgte nicht mit den Augen, reagierte nicht auf Geräusche und hatte keine Kopfkontrolle. Die Ärzte beobachteten sehr niedrigen Muskeltonus, instabile Rumpfbewegungen und schnelle, unkontrollierte Augenbewegungen (Nystagmus). Sein Gesicht zeigte ein langes Gesicht, hohe Stirn, Hautfalten an den inneren Augenwinkeln, lange Lidspalten, eine abgeflachte Nasenwurzel und eine gerundete Nasenspitze mit oberlippenbetonter Länge. Im Laufe des Wachsens konnte er schließlich stehen und einige einfache Silben äußern, blieb aber schwer betroffen und sehr hyperaktiv.
Verborgene Veränderungen in Gehirn, Augen und Nieren
Bildgebende Untersuchungen des Gehirns des Kindes zeigten mehrere strukturelle Abweichungen: Die verbindende Brücke zwischen den Hirnhälften war unterentwickelt, die Myelinisierung der Nervenfasern war verzögert, und tief im Bereich des Nucleus caudatus fand sich eine kleine mit Flüssigkeit gefüllte Zyste. Augenuntersuchungen zeigten Schäden an den Zapfenzellen der Netzhaut, die für scharfes zentrales Sehen und Farbwahrnehmung nötig sind, was seine schlechten visuellen Reaktionen erklärt. Bildgebung des Abdomens ergab eine weitere ungewöhnliche Befundkonstellation: Beide Nieren waren miteinander verwachsen und niedrig im Becken lokalisiert anstatt an ihren üblichen Positionen. Diese Nieren- und Augenprobleme waren in früheren FRA10AC1-Fällen nur selten beschrieben worden, weshalb das Team das gesamte Protein-codierende Genom des Kindes sorgfältig nach anderen plausiblen Ursachen durchsuchte und keine fand.
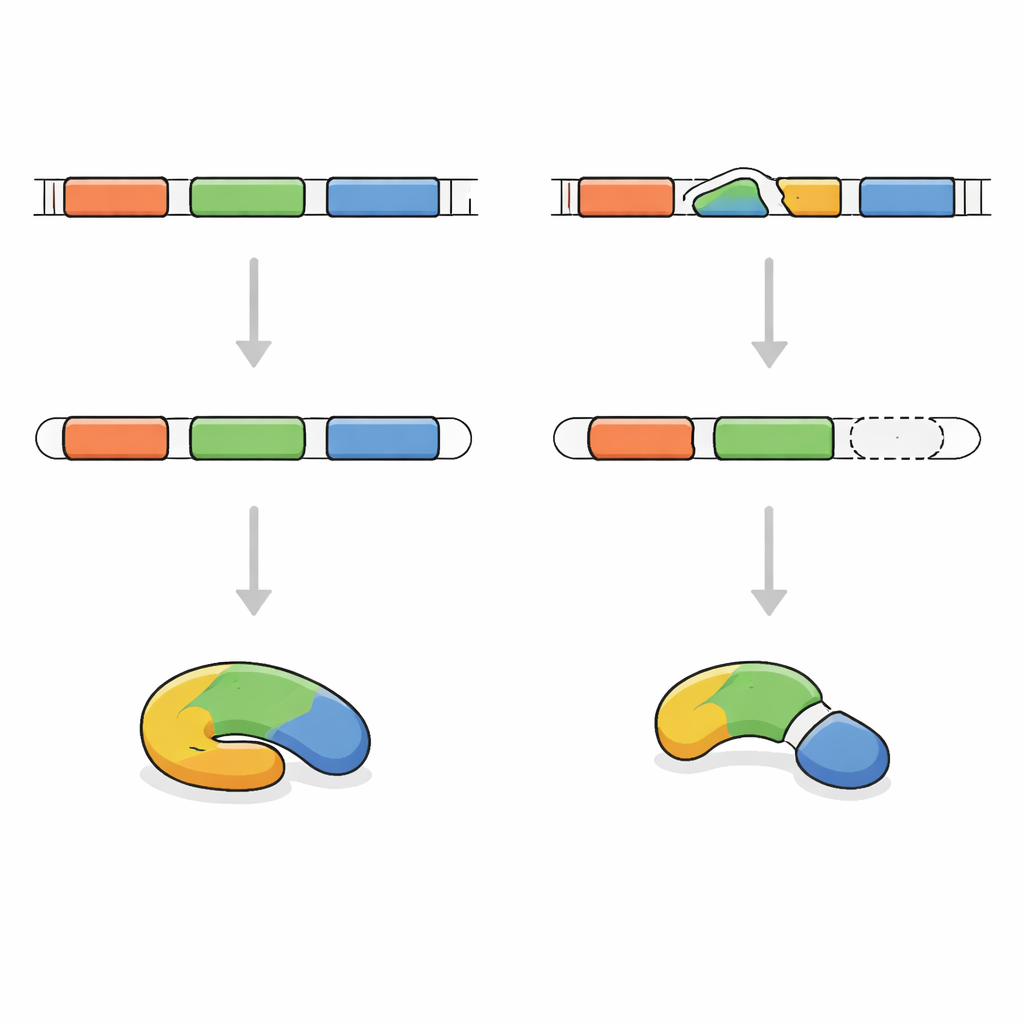
Die fehlerhafte genetische Botschaft verfolgen
Um die Ursache zu ermitteln, sequenzierten die Forschenden die protein-codierenden Abschnitte der DNA des Jungen. Sie entdeckten, dass beide Kopien seines FRA10AC1-Gens dieselbe winzige Veränderung an einem kritischen „Verknüpfungspunkt“ aufwiesen, der während der Nachrichtenbearbeitung genutzt wird. Seine Eltern, die gesund waren, trugen jeweils eine veränderte und eine normale Kopie. Das Team untersuchte anschließend die aus seinen Blutzellen gebildete FRA10AC1-Botschaft. Anstatt alle erwarteten Segmente zu enthalten, fehlte ein Stück vollständig. Dies verschob den Leserahmen, brachte ein frühzeitiges Stoppsignal ins Spiel und führte zu einem verkürzten, vermutlich funktionslosen Protein. Nach internationalen Richtlinien zur Bewertung genetischer Varianten wurde diese Veränderung als eindeutig krankheitsverursachend eingestuft.
Was das für Familien bedeutet
Indem dieser Fall dem kleinen bekannten Kollektiv hinzugefügt wird, trägt die Studie zur Klarstellung des Bildes der FRA10AC1-assoziierten Erkrankung bei. Kinder mit kompletter Verlustfunktion des Proteins, wie der beschriebene Patient, zeigen tendenziell schwerere geistige und motorische Beeinträchtigungen und haben häufiger zusätzliche angeborene Fehler, die Organe wie Herz, Nieren, Augen und Haut betreffen. Dagegen hatten zuvor berichtete Kinder mit einer milderen, teilweise funktionstüchtigen FRA10AC1-Variante weniger schwere Lernprobleme und weniger Organanomalien. Für Familien und Kliniker unterstreicht diese Arbeit, dass eine Störung in diesem einzelnen Gen eine wiedererkennbare Kombination aus Gesichtsmerkmalen, Entwicklungsverzögerungen und Hirnveränderungen verursachen kann und dass sorgfältige Untersuchungen von Nieren und Augen wichtig sind, sobald die Diagnose vermutet wird.
Zitation: Abdel-Hamid, M.S., Abdel-Salam, G.M.H. A novel homozygous splicing variant in FRA10AC1: further delineation of the phenotype. J Hum Genet 71, 363–367 (2026). https://doi.org/10.1038/s10038-025-01447-6
Schlüsselwörter: neuroentwicklungsstörung, Spleißosom, FRA10AC1-Gen, genetische Varianten, Corpus callosum