Clear Sky Science · es
Una nueva variante homocigótica de empalme en FRA10AC1: mayor definición del fenotipo
Cuando la edición genética falla
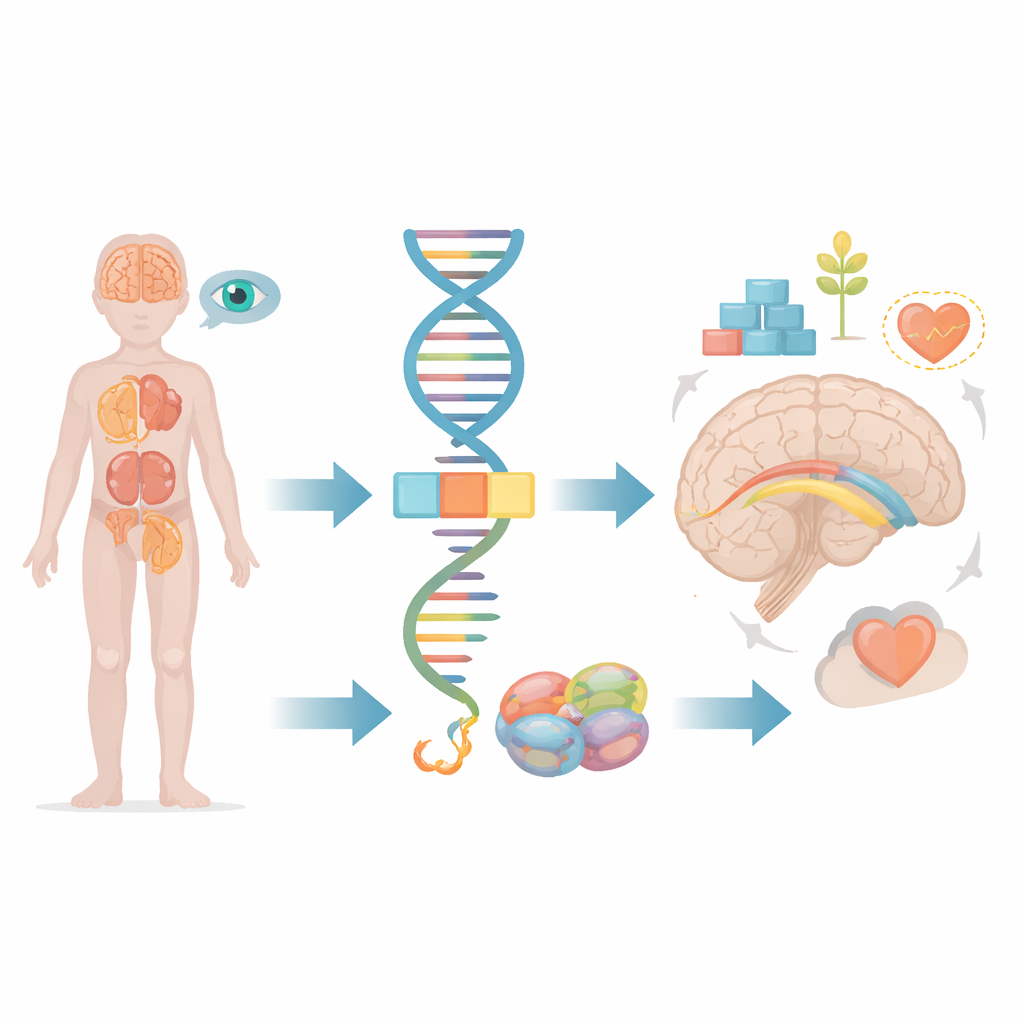
La mayoría de nosotros pensamos en los genes como planos estáticos, pero en realidad nuestras células están constantemente recortando y pegando mensajes genéticos antes de convertirlos en proteínas funcionales. Este artículo explora qué ocurre cuando un pequeño fallo en ese proceso de edición afecta a un gen llamado FRA10AC1, provocando problemas de desarrollo graves en niños. Siguiendo a un paciente joven y comparándolo con los pocos casos conocidos en el mundo, los investigadores muestran cómo un desajuste sutil en el procesamiento de la información genética puede repercutir en el cerebro, los riñones, los ojos y otros órganos.
Los pequeños editores de la célula
Dentro de cada célula, largos tramos de código genético crudo deben ser recortados y unidos antes de poder utilizarse. Esta tarea la realiza una máquina molecular conocida como el espliceosoma, que elimina segmentos no utilizados y cose los útiles. El gen FRA10AC1 produce una proteína auxiliar que se sitúa en el borde de esta máquina y ajusta cómo se efectúan los cortes y las uniones. Estudios previos encontraron que los niños que heredan copias defectuosas de FRA10AC1 de ambos progenitores pueden desarrollar un patrón distintivo de retraso en el desarrollo, rasgos faciales inusuales, crecimiento deficiente y alteraciones en el grueso haz de fibras nerviosas que conecta las dos mitades del cerebro, llamado cuerpo calloso. Sin embargo, solo se habían descrito diez pacientes de este tipo en todo el mundo, lo que dejaba muchas preguntas sobre el espectro completo de síntomas y cómo distintos tipos de fallos en el gen influyen en el resultado.
La historia de un niño aporta nuevas piezas
Los autores describen a un niño egipcio nacido de padres emparentados tras un embarazo sin complicaciones. Desde la primera infancia mostró un retraso marcado en todas las áreas del desarrollo: no sonreía, no seguía con la mirada, no respondía a los sonidos ni había desarrollado control cefálico a los ocho meses de edad. Los médicos observaron tono muscular muy bajo, movimientos inestables del tronco y movimientos oculares rápidos e incontrolados llamados nistagmo. Su aspecto facial incluía cara alargada, frente alta, pliegues de piel en las esquinas internas de los ojos, hendiduras palpebrales largas, puente nasal aplanado y una punta nasal redondeada con un labio superior largo. Al crecer, finalmente consiguió ponerse de pie y pronunciar algunas sílabas simples, pero permaneció con un retraso grave y muy hiperactivo.
Cambios ocultos en cerebro, ojos y riñones
Las exploraciones del cerebro del niño revelaron varias diferencias estructurales: el puente que conecta las dos mitades del cerebro estaba poco desarrollado, la formación del material aislante alrededor de las fibras nerviosas (mielinización) estaba retrasada y había un pequeño quiste lleno de líquido en una región profunda llamada núcleo caudado. Los exámenes oculares mostraron daño en las células cónicas de la retina, necesarias para la visión central nítida y el color, lo que explica sus respuestas visuales pobres. Las imágenes del abdomen revelaron otro hallazgo inusual: ambos riñones estaban fusionados y situados bajos en la pelvis en lugar de sus posiciones habituales. Estos problemas renales y oculares habían aparecido solo raramente en casos previos relacionados con FRA10AC1, por lo que el equipo inspeccionó cuidadosamente todo el conjunto de genes codificadores de proteínas del niño para buscar otras explicaciones plausibles y no encontró ninguna.
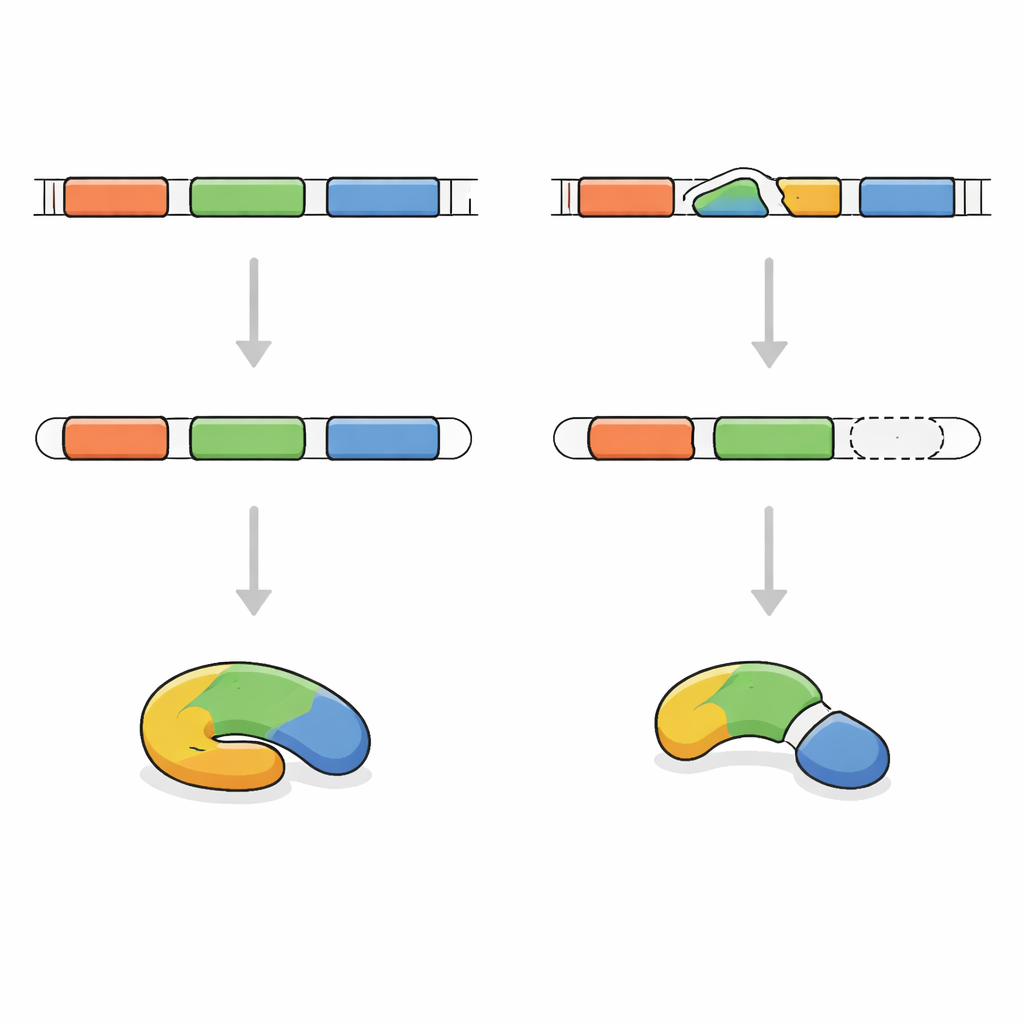
Rastreando el mensaje genético defectuoso
Para identificar la causa, los investigadores secuenciaron las porciones codificantes de proteínas del ADN del niño. Descubrieron que ambas copias de su gen FRA10AC1 presentaban el mismo cambio diminuto en un "punto de unión" crítico usado durante la edición del mensaje. Sus padres, sanos, portaban cada uno una copia alterada y una normal. El equipo examinó entonces el mensaje (ARNm) de FRA10AC1 producido en las células sanguíneas del niño. En lugar de incluir todos los segmentos esperados, el mensaje había omitido por completo una pieza. Esto provocó un cambio en el marco de lectura, introduciendo una señal de parada temprana y conduciendo a una proteína acortada y probablemente inútil. Basándose en las guías internacionales para la evaluación de variantes genéticas, este cambio se clasificó como claramente patogénico.
Qué significa esto para las familias
Al sumar el caso de este niño al reducido número ya conocido, el estudio ayuda a aclarar el panorama del trastorno relacionado con FRA10AC1. Los niños con pérdida completa de la proteína, como este paciente, tienden a presentar mayores dificultades intelectuales y motoras y son más propensos a tener defectos congénitos adicionales que afectan órganos como el corazón, los riñones, los ojos y la piel. En contraste, los niños descritos previamente con una versión más leve y parcialmente funcional de FRA10AC1 mostraron problemas de aprendizaje menos severos y menos anomalías orgánicas. Para las familias y los clínicos, este trabajo subraya que una alteración en este único gen puede explicar una combinación reconocible de rasgos faciales, retrasos en el desarrollo y cambios cerebrales, y que las exploraciones cuidadosas de riñones y ojos son importantes una vez que se sospecha el diagnóstico.
Cita: Abdel-Hamid, M.S., Abdel-Salam, G.M.H. A novel homozygous splicing variant in FRA10AC1: further delineation of the phenotype. J Hum Genet 71, 363–367 (2026). https://doi.org/10.1038/s10038-025-01447-6
Palabras clave: trastorno del neurodesarrollo, espliceosoma, gen FRA10AC1, variantes genéticas, Cuerpo calloso