Clear Sky Science · pl
Nowy homozygotyczny wariant splicingowy w FRA10AC1: dalsze doprecyzowanie fenotypu
Kiedy edycja genów idzie nie tak
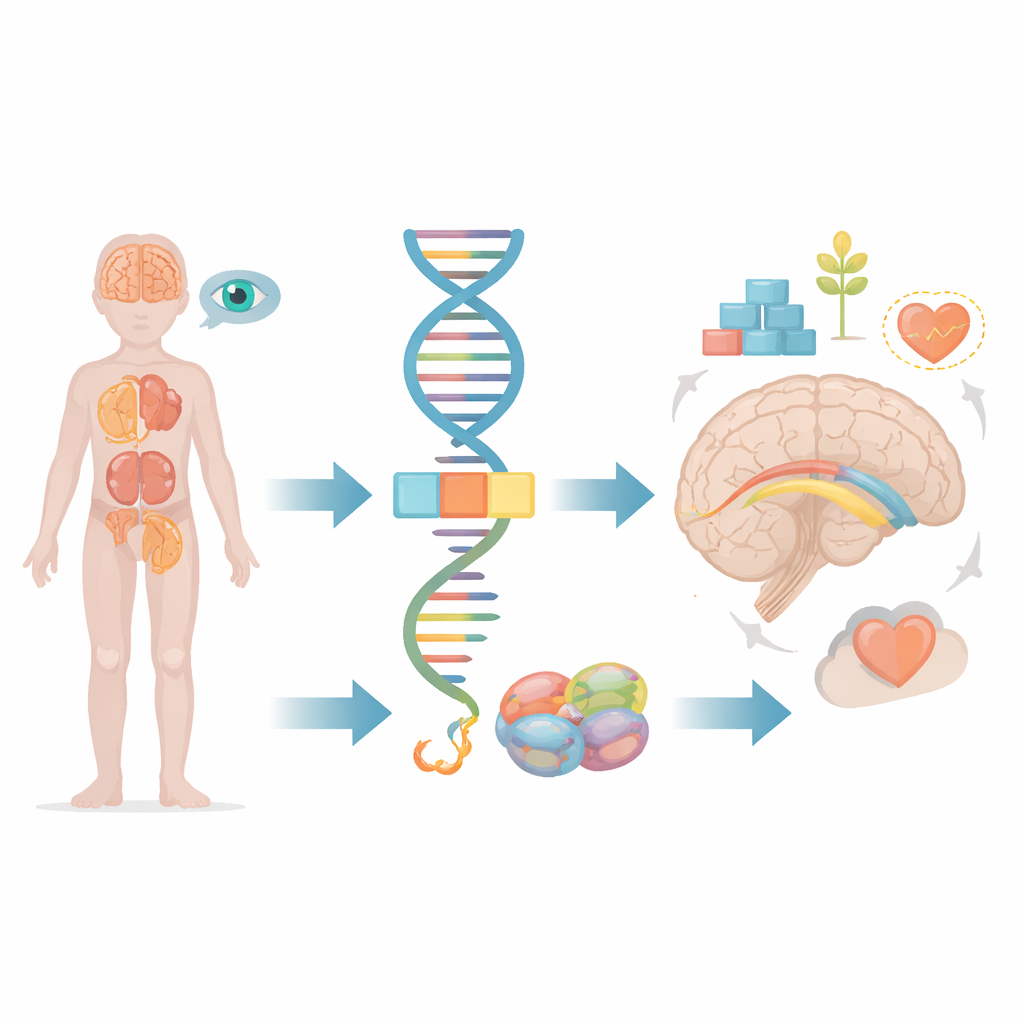
Większość z nas myśli o genach jak o statycznych planach, tymczasem w rzeczywistości nasze komórki nieustannie wycinają i sklejają komunikaty genetyczne, zanim przekształcą je w działające białka. Artykuł bada, co się dzieje, gdy drobny błąd w tym procesie edycji dotyczy genu o nazwie FRA10AC1, prowadząc do poważnych zaburzeń rozwojowych u dzieci. Na przykładzie jednego młodego pacjenta i porównaniu z nielicznymi znanymi przypadkami na świecie autorzy pokazują, jak subtelna usterka w przetwarzaniu informacji genetycznej może odbić się na mózgu, nerkach, oczach i innych narządach.
Maleńcy redaktorzy komórki
W każdej komórce długie fragmenty surowego kodu genetycznego muszą zostać przycięte i połączone, zanim będzie można je wykorzystać. To zadanie wykonuje maszyneria molekularna zwana spliceosomem, która wycina nieużywane segmenty i zszywa te przydatne. Gen FRA10AC1 koduje białko pomocnicze, które znajduje się na obrzeżach tej machiny i dopracowuje sposób, w jaki następuje cięcie i łączenie. Wcześniejsze badania wykazały, że dzieci, które odziedziczyły wadliwe kopie FRA10AC1 od obojga rodziców, mogą rozwijać charakterystyczny zestaw opóźnień rozwojowych, nietypowe rysy twarzy, słaby wzrost oraz zmiany w grubym pęku włókien nerwowych łączącym dwie półkule mózgu, zwanym ciałem modzelowatym. Jednak opisano do tej pory tylko dziesięciu takich pacjentów na świecie, co pozostawia wiele pytań dotyczących pełnego zakresu objawów i tego, jak różne typy uszkodzeń genu wpływają na przebieg choroby.
Historia jednego dziecka dodaje nowe elementy
Autorzy opisują egipskiego chłopca urodzonego przez spokrewnionych rodziców po prawidłowej ciąży. Już we wczesnym niemowlęctwie wykazywał wyraźne opóźnienia we wszystkich sferach rozwoju: nie uśmiechał się, nie śledził wzrokiem, nie reagował na dźwięki ani nie osiągnął kontroli głowy w wieku ośmiu miesięcy. Lekarze zauważyli bardzo niskie napięcie mięśniowe, niestabilne ruchy tułowia oraz szybkie, mimowolne ruchy oczu zwane oczopląsem. Wygląd twarzy obejmował długą twarz, wysokie czoło, fałdy skóry w wewnętrznych kącikach oczu, długie szpary powiek, spłaszczony grzbiet nosa oraz zaokrągloną końcówkę nosa i długą górną wargę. W miarę dorastania zdołał w końcu stanąć i wypowiedzieć kilka prostych sylab, lecz pozostawał ciężko opóźniony i bardzo nadpobudliwy.
Ukryte zmiany w mózgu, oczach i nerkach
Badania obrazowe mózgu dziecka wykazały kilka nieprawidłowości strukturalnych: most łączący dwie półkule był niedorozwinięty, izolacja włókien nerwowych (osłonka mielinowa) była opóźniona w tworzeniu, a głęboko w obszarze nazywanym jądrem ogoniastym obecna była mała torbiel wypełniona płynem. Badanie oczu ujawniło uszkodzenie komórek stożkowych siatkówki, niezbędnych do ostrego widzenia centralnego i rozpoznawania kolorów, co tłumaczyło słabe reakcje wzrokowe. Obrazowanie jamy brzusznej wykazało kolejny nietypowy objaw: obie nerki były zrosłe i leżały nisko w miednicy zamiast w zwykłych miejscach. Problemy z nerkami i oczami rzadko pojawiały się w wcześniejszych przypadkach związanych z FRA10AC1, więc zespół dokładnie przeanalizował cały zestaw genów kodujących białka u dziecka, by poszukać innych możliwych wyjaśnień — nie znaleziono żadnych.
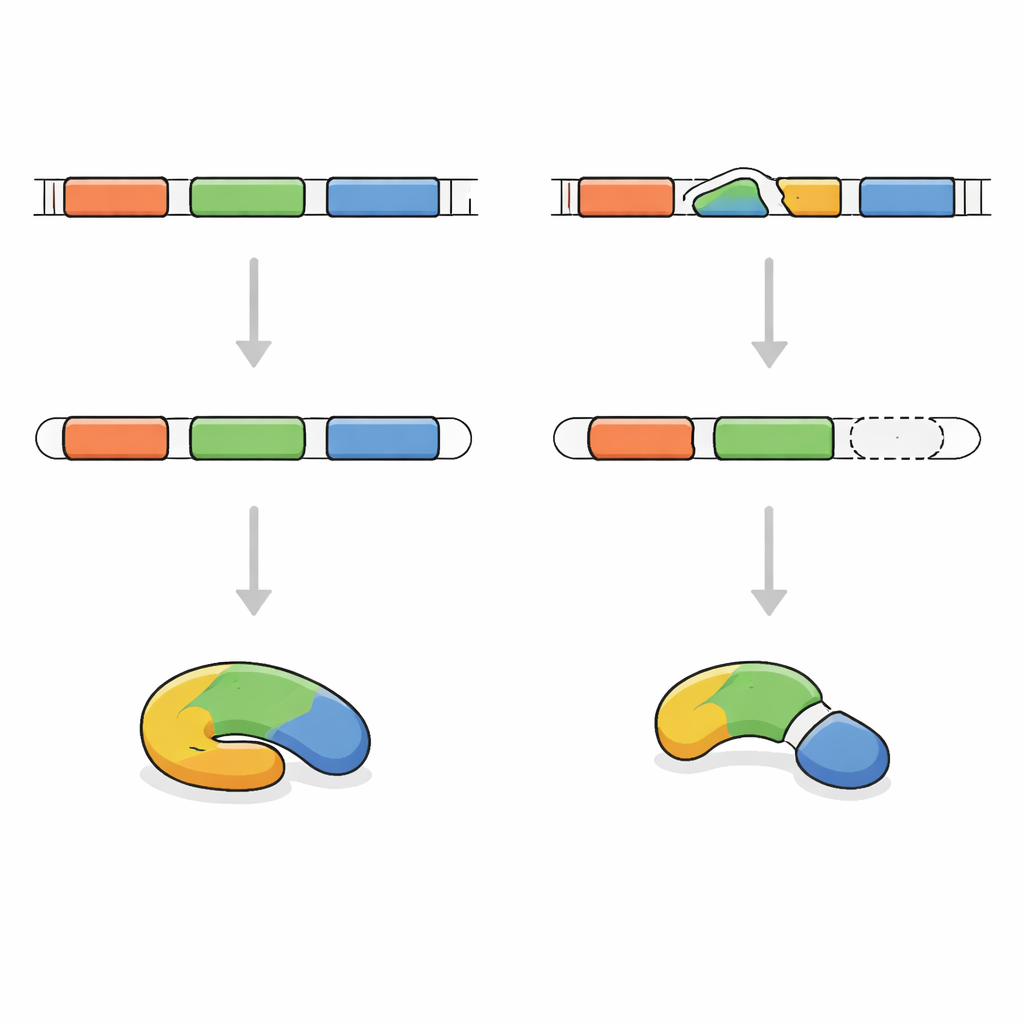
Śledzenie wadliwego komunikatu genetycznego
Aby ustalić przyczynę, badacze zsekwencjonowali odcinki DNA odpowiadające za kodowanie białek u chłopca. Odkryli, że obie kopie jego genu FRA10AC1 miały tę samą drobną zmianę w krytycznym „punkcie łączenia” używanym podczas edycji komunikatu. Rodzice, którzy byli zdrowi, każdy nosił jedną zmienioną i jedną prawidłową kopię. Zespół następnie przeanalizował transkrypt FRA10AC1 pochodzący z jego krwi. Zamiast zawierać wszystkie oczekiwane segmenty, komunikat pominął jeden fragment całkowicie. Spowodowało to przesunięcie ramki odczytu, wprowadzenie wczesnego sygnału stop i powstanie skróconego, prawdopodobnie nieaktywnego białka. Na podstawie międzynarodowych wytycznych dotyczących oceny wariantów genetycznych tę zmianę zaklasyfikowano jako jednoznacznie chorobotwórczą.
Co to oznacza dla rodzin
Dodając opis tego chłopca do niewielkiej liczby już znanych przypadków, badanie pomaga wyjaśnić obraz zaburzenia związanego z FRA10AC1. Dzieci z całkowitą utratą funkcji białka, jak ten pacjent, mają zwykle cięższe trudności intelektualne i motoryczne oraz częściej występują u nich dodatkowe wady wrodzone dotyczące narządów takich jak serce, nerki, oczy i skóra. Natomiast wcześniej opisywane dzieci z łagodniejszą, częściowo funkcjonalną wersją FRA10AC1 miały mniej nasilone problemy z nauką i mniej anomalii narządowych. Dla rodzin i klinicystów praca ta podkreśla, że wada pojedynczego genu może leżeć u podstaw rozpoznawalnego zespołu cech twarzy, opóźnień rozwojowych i zmian mózgowych, oraz że po podejrzeniu rozpoznania ważne są dokładne badania nerek i oczu.
Cytowanie: Abdel-Hamid, M.S., Abdel-Salam, G.M.H. A novel homozygous splicing variant in FRA10AC1: further delineation of the phenotype. J Hum Genet 71, 363–367 (2026). https://doi.org/10.1038/s10038-025-01447-6
Słowa kluczowe: zaburzenie neurorozwojowe, spliceosom, gen FRA10AC1, warianty genetyczne, ciało modzelowate