Clear Sky Science · pt
Acelerando formulações farmacêuticas em fluido supercrítico por meio de predição interpretável e orientada por dados da solubilidade de fármacos
Por que isso importa para os medicamentos do futuro
Muitos medicamentos promissores nunca chegam aos pacientes porque simplesmente não se dissolvem bem o suficiente para o organismo absorvê-los. O estudo por trás deste artigo explora como prever, por computador, com que facilidade diferentes fármacos se dissolvem em uma forma especial de dióxido de carbono conhecida como fluido supercrítico. Ao usar ferramentas modernas orientadas por dados, os autores buscam reduzir tentativas e erros laboratoriais custosos e acelerar a jornada da molécula até o medicamento.
Uma forma mais limpa de produzir fármacos de difícil dissolução
O dióxido de carbono supercrítico comporta-se tanto como líquido quanto como gás, conferindo-lhe um poder incomum para transportar e moldar moléculas de fármacos. Ele pode ajudar a criar partículas minúsculas, incorporar fármacos em sistemas de liberação e fazê-lo sem deixar solventes tóxicos. No entanto, cada fármaco responde de modo diferente: variações de pressão, temperatura e a própria estrutura do fármaco podem alterar dramaticamente quanto dele se dissolve. Medir esses efeitos para cada novo composto em equipamentos de alta pressão é lento e caro, de modo que ser capaz de prever a solubilidade a partir de dados é altamente atraente para cientistas de formulação e fabricantes de medicamentos.
Construindo um motor de predição inteligente
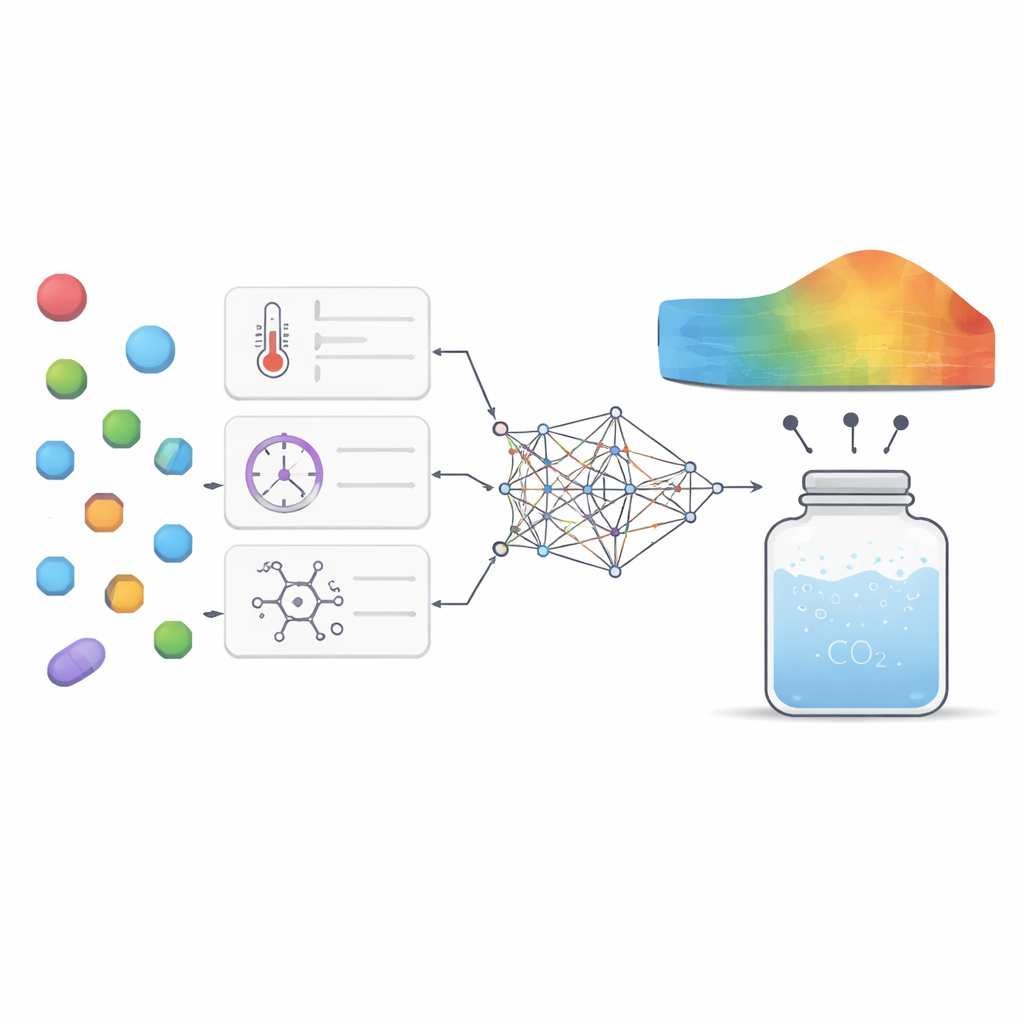
Para enfrentar esse desafio, os pesquisadores reuniram 252 medições de alta qualidade de experimentos anteriores com sete fármacos muito diferentes, que vão de medicamentos para úlcera e epilepsia a antifúngicos e antivirais. Para cada ponto de dado coletaram condições básicas como temperatura e pressão, junto com propriedades simples do fármaco, como massa molecular e ponto de fusão. Em seguida, treinaram vários modelos de aprendizado de máquina para aprender a relação entre essas entradas e quanto de cada fármaco se dissolvia no dióxido de carbono supercrítico. Foram usados dois tipos principais de modelos: regressão por vetores de suporte, que tenta ajustar uma curva suave aos dados, e extreme gradient boosting, um método avançado baseado em árvores que combina muitas pequenas árvores de decisão em um preditor forte.
Deixando algoritmos de busca inspirados na natureza ajustar os modelos
Escolher as configurações internas desses modelos, conhecidas como hiperparâmetros, pode afetar muito seu desempenho. Em vez de chutar ou usar buscas em grade simples, a equipe pegou emprestado ideias do comportamento animal. Aplicaram dois métodos de otimização “bio-inspirados”, um baseado na maneira como gansos-comuns buscam alimento e migram, e outro inspirado nas táticas de sobrevivência de lagartos com chifres. Esses algoritmos exploram muitas combinações de configurações de modelo e gradualmente convergem para as que fornecem as previsões mais precisas com menor erro. Ao enquadrar isso como um problema de múltiplos objetivos, os autores equilibraram duas metas ao mesmo tempo: minimizar o erro de predição e maximizar quanto da variação nos dados o modelo explica, mantendo também atenção ao tempo de computação.
Vendo quais alavancas importam mais
Além da precisão bruta, os autores queriam que o modelo oferecesse insight científico em vez de ser uma “caixa-preta” misteriosa. Usaram duas ferramentas de análise complementares para perguntar quão fortemente cada fator de entrada moldava a saída do modelo e como esses fatores interagiam. Ambas as técnicas destacaram pressão e massa molecular como as alavancas mais importantes: pressão mais alta geralmente aumentava a solubilidade ao compactar o dióxido de carbono, enquanto moléculas mais pesadas tendiam a se dissolver com menos facilidade. Temperatura e ponto de fusão tiveram papéis mais sutis, mas ainda significativos, atuando principalmente em interação com a pressão e refletindo o quão firmemente a estrutura cristalina do fármaco se mantém. O modelo também reproduziu padrões termodinâmicos conhecidos, como o fato de que variar a temperatura pode tanto aumentar quanto diminuir o efeito da pressão dependendo da faixa, o que dá confiança de que estava capturando comportamento físico real em vez de apenas memorizar números.
O que os resultados significam para o desenvolvimento de fármacos
O modelo híbrido de melhor desempenho, que combinou a abordagem de gradient boosting com o otimizador inspirado no lagarto com chifres, reduziu o erro de predição em cerca de 40% em comparação com uma linha de base não ajustada. Ele casou bem não apenas com os valores médios de solubilidade observados nos experimentos, mas também com sua dispersão e com casos raros de alta solubilidade. Esse nível de acurácia significa que o modelo pode atuar como uma poderosa ferramenta de pré-seleção: pesquisadores podem explorar faixas de pressão e temperatura em um laptop antes de se comprometerem com experimentos em alta pressão, reduzindo esforço desperdiçado em candidatos ruins. Embora a abordagem ainda dependa de um conjunto de dados modesto e de um conjunto limitado de propriedades de fármacos, e a versão mais precisa seja exigente em termos computacionais, a estrutura mostra como aprendizado de máquina interpretável pode tanto acelerar formulações sem solventes quanto aprofundar nossa compreensão de por que alguns fármacos se dissolvem melhor que outros em dióxido de carbono supercrítico.
Citação: Khafagy, ES., Lila, A.S.A. & Pishnamazi, M. Accelerating supercritical pharmaceutical formulation via interpretable data-driven prediction of drug solubility. Sci Rep 16, 11006 (2026). https://doi.org/10.1038/s41598-026-44161-9
Palavras-chave: solubilidade de fármacos, dióxido de carbono supercrítico, aprendizado de máquina, formulação farmacêutica, modelagem orientada por dados