Clear Sky Science · en
Accelerating supercritical pharmaceutical formulation via interpretable data-driven prediction of drug solubility
Why this matters for future medicines
Many promising medicines never reach patients because they simply do not dissolve well enough for the body to absorb them. The study behind this article explores how to predict, on a computer, how easily different drugs dissolve in a special form of carbon dioxide known as a supercritical fluid. By using modern data-driven tools, the authors aim to cut down on costly laboratory trial-and-error and speed up the journey from molecule to medicine.
A cleaner way to make hard-to-dissolve drugs
Supercritical carbon dioxide behaves like both a liquid and a gas, giving it unusual power to carry and shape drug molecules. It can help create tiny particles, load drugs into delivery systems, and do so without leaving behind toxic solvents. However, every drug responds differently: changes in pressure, temperature, and the drug’s own structure can dramatically alter how much of it dissolves. Measuring these effects for each new compound in high-pressure equipment is slow and expensive, so being able to predict solubility from data is highly attractive for formulation scientists and drug makers.
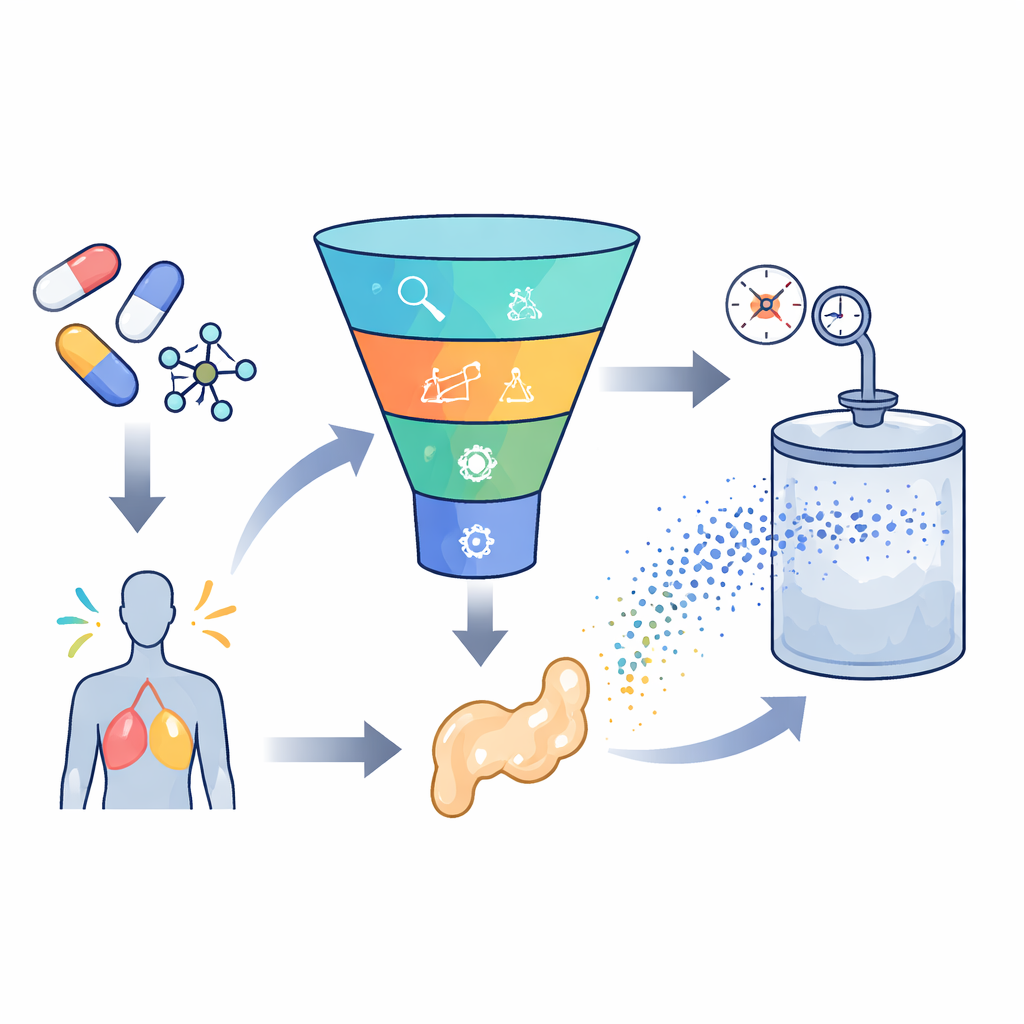
Building a smart prediction engine
To tackle this challenge, the researchers gathered 252 high-quality measurements from previous experiments on seven very different drugs, ranging from ulcer and epilepsy medicines to antifungals and antivirals. For each data point they collected basic conditions such as temperature and pressure, along with simple drug properties like molecular weight and melting point. They then trained several machine-learning models to learn the link between these inputs and how much of each drug dissolved in supercritical carbon dioxide. Two main types of models were used: support vector regression, which tries to draw a smooth curve through the data, and extreme gradient boosting, an advanced tree-based method that combines many small decision trees into one strong predictor.
Letting nature-inspired search algorithms tune the models
Choosing the internal settings of these models, known as hyperparameters, can greatly affect how well they perform. Instead of guessing or using simple grid searches, the team borrowed ideas from animal behavior. They applied two “bio-inspired” optimization methods, one based on the way greylag geese forage and migrate, and another inspired by horned lizards’ survival tactics. These algorithms explore many combinations of model settings and gradually move toward those that give the most accurate predictions with the least error. By framing this as a multi-objective problem, the authors balanced two goals at once: minimizing prediction error and maximizing how much of the variation in the data the model explains, while also keeping an eye on computing time.
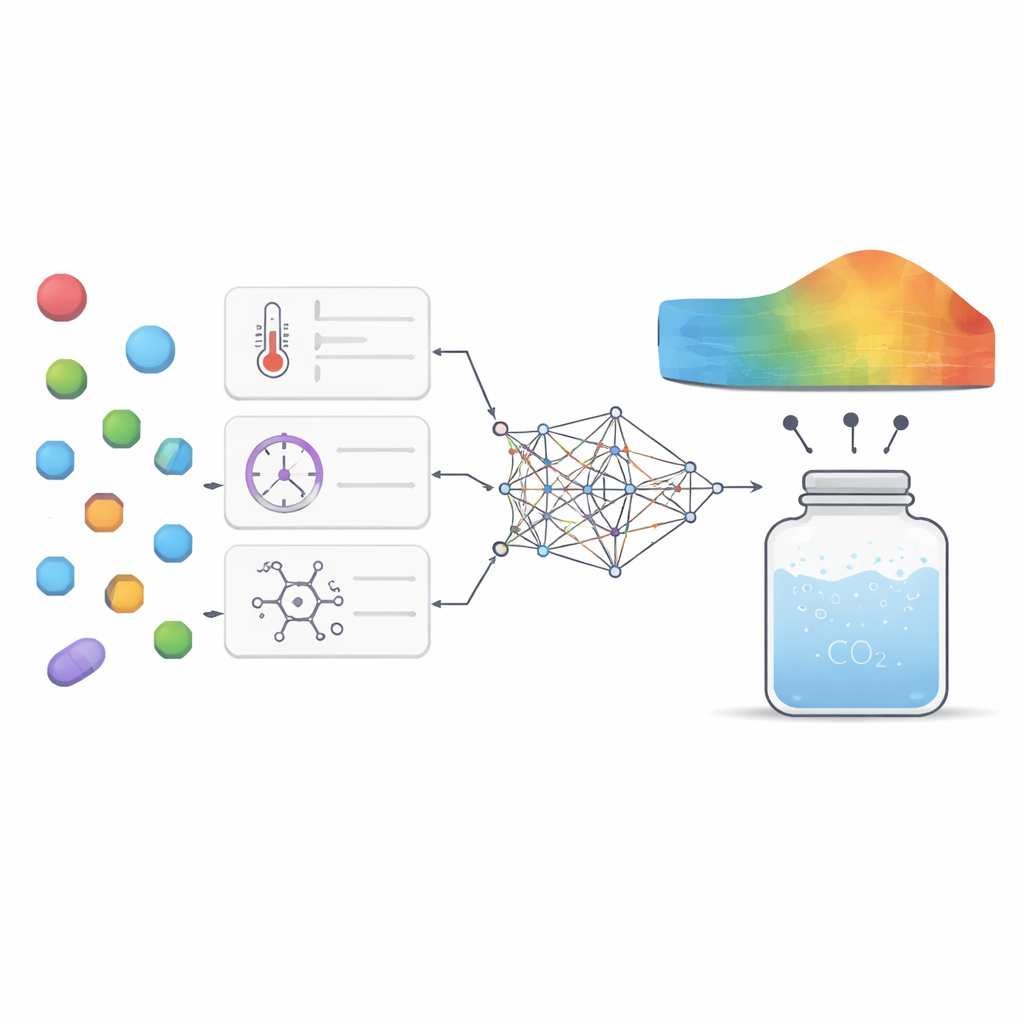
Seeing which levers matter most
Beyond raw accuracy, the authors wanted the model to offer scientific insight rather than be a mysterious “black box.” They used two complementary analysis tools to ask how strongly each input factor shaped the model’s output and how those factors interacted. Both techniques highlighted pressure and molecular weight as the most important levers: higher pressure generally increased solubility by packing carbon dioxide more tightly, while heavier molecules tended to dissolve less easily. Temperature and melting point played subtler but still meaningful roles, mainly by interacting with pressure and reflecting how tightly the drug’s crystal structure holds together. The model also reproduced known thermodynamic patterns, such as how changing temperature can either enhance or weaken the effect of pressure depending on the range, lending confidence that it was capturing real physical behavior rather than just memorizing numbers.
What the results mean for drug development
The best-performing hybrid model, which combined the gradient-boosting approach with the horned lizard optimizer, cut prediction error by roughly 40 percent compared with an untuned baseline. It closely matched not just the average solubility values observed in experiments, but also their spread and rare high-solubility cases. This level of accuracy means the model can act as a powerful pre-screening tool: researchers can explore pressure and temperature ranges on a laptop before committing to high-pressure experiments, reducing wasted effort on poor candidates. Although the approach still depends on a modest dataset and a limited set of drug properties, and the most accurate version is computationally demanding, the framework shows how interpretable machine learning can both speed up solvent-free formulations and deepen our understanding of why some drugs dissolve better than others in supercritical carbon dioxide.
Citation: Khafagy, ES., Lila, A.S.A. & Pishnamazi, M. Accelerating supercritical pharmaceutical formulation via interpretable data-driven prediction of drug solubility. Sci Rep 16, 11006 (2026). https://doi.org/10.1038/s41598-026-44161-9
Keywords: drug solubility, supercritical carbon dioxide, machine learning, pharmaceutical formulation, data-driven modeling