Clear Sky Science · es
Aceleración de la formulación farmacéutica en condiciones supercríticas mediante predicción interpretable y basada en datos de la solubilidad de fármacos
Por qué importa para las medicinas del futuro
Muchas medicinas prometedoras nunca llegan a los pacientes porque simplemente no se disuelven lo suficiente como para que el cuerpo las absorba. El estudio que respalda este artículo explora cómo predecir, por ordenador, qué tan fácilmente diferentes fármacos se disuelven en una forma especial de dióxido de carbono conocida como fluido supercrítico. Al emplear herramientas modernas basadas en datos, los autores pretenden reducir los costosos ensayos experimentales por prueba y error y acelerar el trayecto de la molécula al medicamento.
Una forma más limpia de fabricar fármacos de difícil disolución
El dióxido de carbono supercrítico se comporta tanto como líquido como gas, lo que le confiere una capacidad inusual para transportar y estructurar moléculas de fármacos. Puede ayudar a crear partículas diminutas, cargar fármacos en sistemas de liberación y hacerlo sin dejar disolventes tóxicos. Sin embargo, cada fármaco responde de manera diferente: cambios en la presión, la temperatura y la propia estructura del fármaco pueden alterar drásticamente la cantidad que se disuelve. Medir estos efectos para cada compuesto nuevo en equipos de alta presión es lento y caro, por lo que poder predecir la solubilidad a partir de datos resulta muy atractivo para los científicos de formulación y los fabricantes de fármacos.
Construir un motor de predicción inteligente
Para abordar este desafío, los investigadores reunieron 252 mediciones de alta calidad procedentes de experimentos previos sobre siete fármacos muy diferentes, que van desde medicamentos para úlceras y epilepsia hasta antifúngicos y antivirales. Para cada punto de datos recogieron condiciones básicas como temperatura y presión, junto con propiedades sencillas del fármaco como peso molecular y punto de fusión. A continuación entrenaron varios modelos de aprendizaje automático para aprender la relación entre estas entradas y la cantidad de cada fármaco que se disolvía en dióxido de carbono supercrítico. Se emplearon dos tipos principales de modelos: regresión por vectores de soporte, que intenta trazar una curva suave a través de los datos, y extreme gradient boosting, un método avanzado basado en árboles que combina muchos pequeños árboles de decisión en un predictor robusto.
Dejar que algoritmos de búsqueda inspirados en la naturaleza ajusten los modelos
Elegir los parámetros internos de estos modelos, conocidos como hiperparámetros, puede afectar en gran medida a su rendimiento. En lugar de adivinar o usar búsquedas en cuadrícula simples, el equipo tomó ideas del comportamiento animal. Aplicaron dos métodos de optimización “bioinspirados”, uno basado en la forma en que las ocas grises forrajean y migran, y otro inspirado en las tácticas de supervivencia de los lagartos cornudos. Estos algoritmos exploran muchas combinaciones de ajustes del modelo y avanzan gradualmente hacia aquellas que ofrecen las predicciones más precisas con el menor error. Al plantearlo como un problema de múltiples objetivos, los autores equilibraron dos metas a la vez: minimizar el error de predicción y maximizar la proporción de variación de los datos explicada por el modelo, sin perder de vista el tiempo de cálculo.
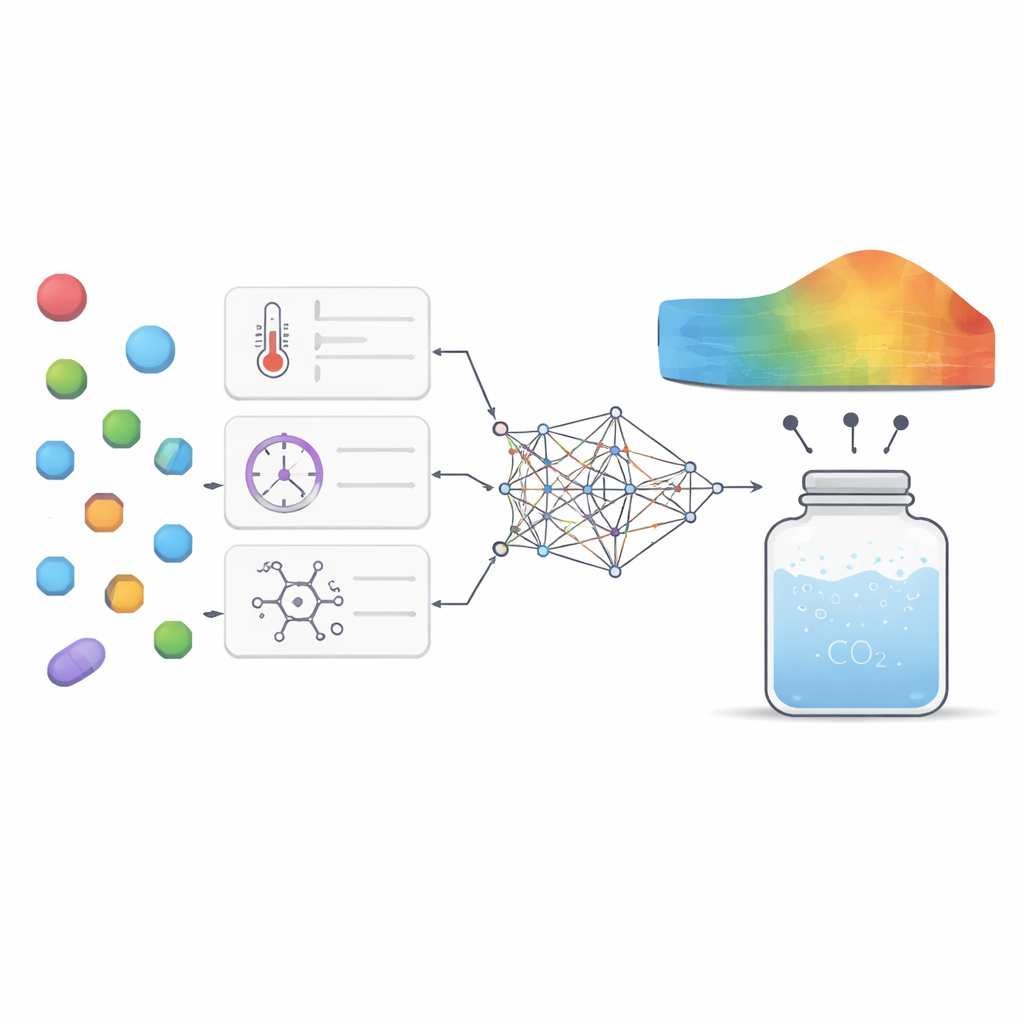
Ver qué palancas importan más
Más allá de la precisión bruta, los autores querían que el modelo ofreciera conocimientos científicos en lugar de ser una caja negra misteriosa. Utilizaron dos herramientas de análisis complementarias para evaluar con qué intensidad cada factor de entrada influía en la salida del modelo y cómo interactuaban esos factores. Ambas técnicas destacaron la presión y el peso molecular como las palancas más importantes: una mayor presión generalmente aumentaba la solubilidad al compactar más el dióxido de carbono, mientras que las moléculas más pesadas tendían a disolverse con menos facilidad. La temperatura y el punto de fusión desempeñaron papeles más sutiles pero aún significativos, principalmente a través de interacciones con la presión y reflejando cuán fuertemente está ligada la estructura cristalina del fármaco. El modelo también reprodujo patrones termodinámicos conocidos, como que el cambio de temperatura puede fortalecer o debilitar el efecto de la presión según el rango, lo que da confianza en que capturaba un comportamiento físico real en lugar de limitarse a memorizar números.
Qué significan los resultados para el desarrollo de fármacos
El modelo híbrido de mejor rendimiento, que combinó el enfoque de gradient boosting con el optimizador inspirado en el lagarto cornudo, redujo el error de predicción en aproximadamente un 40 por ciento en comparación con una línea base sin ajuste. Coincidía estrechamente no solo con los valores medios de solubilidad observados en los experimentos, sino también con su dispersión y con los casos raros de alta solubilidad. Este nivel de precisión convierte al modelo en una potente herramienta de preselección: los investigadores pueden explorar rangos de presión y temperatura en un portátil antes de comprometerse con experimentos a alta presión, reduciendo el esfuerzo desperdiciado en candidatos pobres. Aunque el enfoque aún depende de un conjunto de datos modesto y de un conjunto limitado de propiedades del fármaco, y la versión más precisa exige mucho cálculo, el marco muestra cómo el aprendizaje automático interpretable puede acelerar formulaciones sin disolventes y profundizar nuestra comprensión de por qué algunos fármacos se disuelven mejor que otros en dióxido de carbono supercrítico.
Cita: Khafagy, ES., Lila, A.S.A. & Pishnamazi, M. Accelerating supercritical pharmaceutical formulation via interpretable data-driven prediction of drug solubility. Sci Rep 16, 11006 (2026). https://doi.org/10.1038/s41598-026-44161-9
Palabras clave: solubilidad de fármacos, dióxido de carbono supercrítico, aprendizaje automático, formulación farmacéutica, modelado basado en datos