Clear Sky Science · de
Beschleunigung superkritischer pharmazeutischer Formulierungen durch interpretierbare datengestützte Vorhersage der Arzneimittel-Löslichkeit
Warum das für zukünftige Medikamente wichtig ist
Viele vielversprechende Arzneimittel erreichen Patienten nie, weil sie sich nicht gut genug lösen, um vom Körper aufgenommen zu werden. Die Studie hinter diesem Artikel untersucht, wie man rechnerisch vorhersagen kann, wie leicht sich verschiedene Wirkstoffe in einer speziellen Form von Kohlendioxid — einem superkritischen Fluid — lösen. Mit modernen datengestützten Werkzeugen wollen die Autoren kostspieliges Labor-Probieren-von-Fehlern reduzieren und die Reise vom Molekül zum Medikament beschleunigen.
Ein saubererer Weg, schwer lösliche Wirkstoffe herzustellen
Superkritisches Kohlendioxid verhält sich zugleich wie Flüssigkeit und Gas und besitzt dadurch ungewöhnliche Fähigkeiten, Wirkstoffmoleküle zu transportieren und zu formen. Es kann helfen, winzige Partikel zu erzeugen, Wirkstoffe in Trägersysteme zu laden, und das alles ohne toxische Lösungsmittel zurückzulassen. Jeder Wirkstoff reagiert jedoch anders: Änderungen von Druck, Temperatur und der molekularen Struktur können die Löslichkeit stark beeinflussen. Diese Effekte für jede neue Verbindung in Hochdruckgeräten zu messen ist langsam und teuer, sodass die Vorhersage der Löslichkeit aus Daten für Formulierungswissenschaftler und Arzneimittelhersteller sehr attraktiv ist.
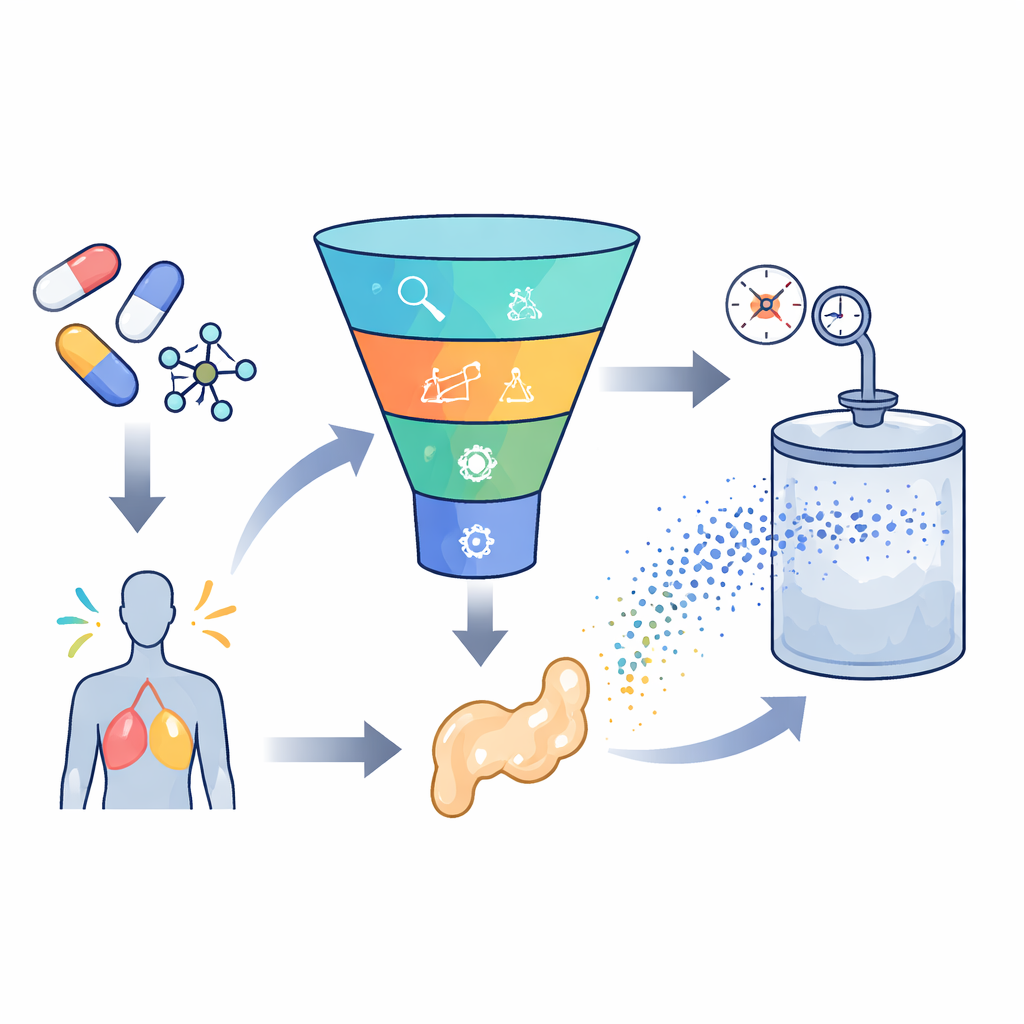
Aufbau einer intelligenten Vorhersage-Engine
Um diese Herausforderung zu meistern, sammelten die Forscher 252 hochwertige Messwerte aus früheren Experimenten an sieben sehr unterschiedlichen Wirkstoffen, von Magenschutz- und Epilepsiemitteln bis zu Antimykotika und Virostatika. Für jeden Datenpunkt erfassten sie grundlegende Bedingungen wie Temperatur und Druck sowie einfache Wirkstoffeigenschaften wie Molekulargewicht und Schmelzpunkt. Anschließend trainierten sie mehrere Modelle des maschinellen Lernens, um die Verbindung zwischen diesen Eingaben und der Löslichkeit jedes Wirkstoffs in superkritischem Kohlendioxid zu erlernen. Es wurden zwei Hauptmodelltypen verwendet: Support-Vector-Regression, die versucht, eine glatte Kurve durch die Daten zu ziehen, und Extreme Gradient Boosting, eine fortgeschrittene baumbasierte Methode, die viele kleine Entscheidungsbäume zu einem starken Prädiktor kombiniert.
Naturinspirierte Suchalgorithmen zur Feinabstimmung der Modelle
Die Wahl der internen Einstellungen dieser Modelle, bekannt als Hyperparameter, kann die Leistung stark beeinflussen. Anstatt zu raten oder einfache Gittersuche zu verwenden, entlehnte das Team Ideen aus dem Tierverhalten. Sie setzten zwei „bio-inspirierte“ Optimierungsmethoden ein: eine basierend auf dem Nahrungs- und Zugverhalten der Graugänse und eine andere, inspiriert von den Überlebensstrategien gehörnter Eidechsen. Diese Algorithmen erkunden viele Kombinationen von Modellparametern und bewegen sich schrittweise zu denen, die die genauesten Vorhersagen mit dem geringsten Fehler liefern. Indem sie dies als Multi-Objective-Problem formulierten, balancierten die Autoren zwei Ziele gleichzeitig: Minimierung des Vorhersagefehlers und Maximierung des erklärten Datenvarianzanteils, wobei auch die Rechenzeit berücksichtigt wurde.
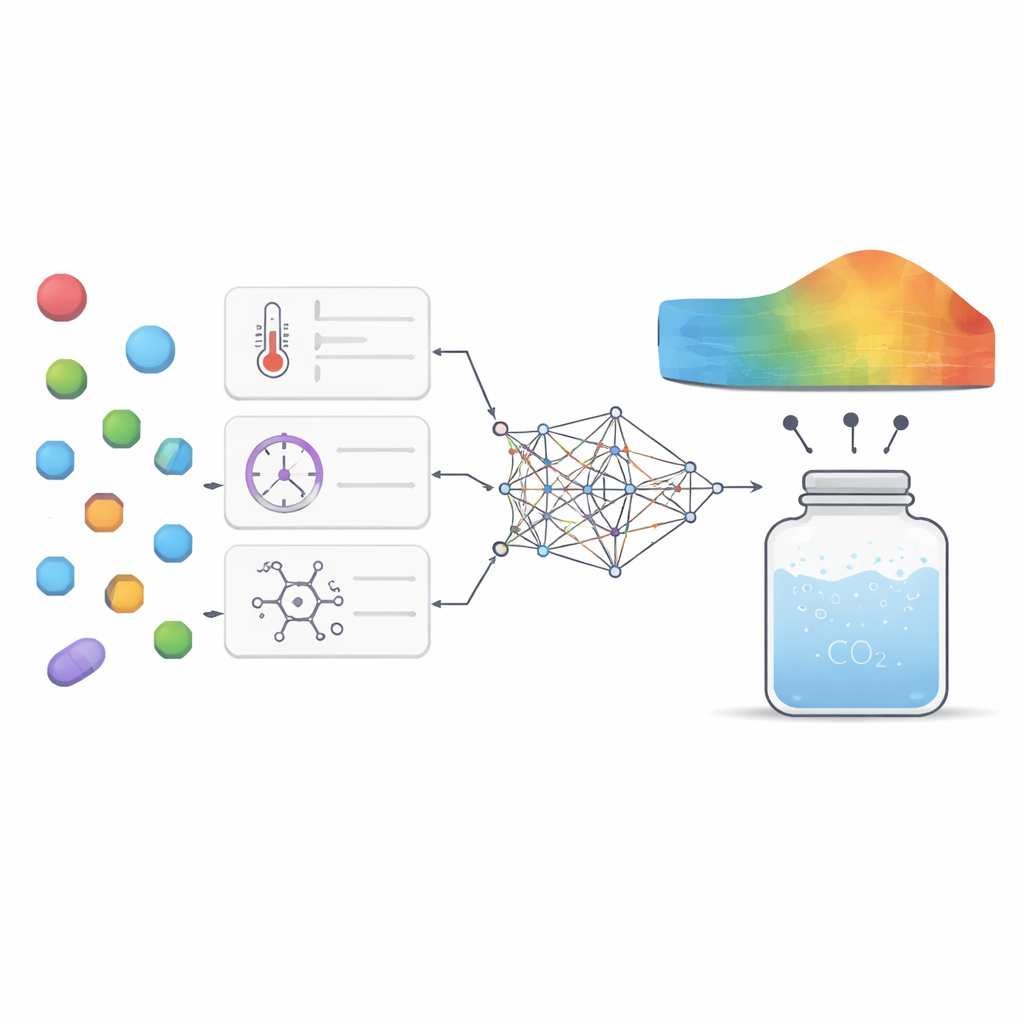
Erkennen, welche Stellschrauben am wichtigsten sind
Über die reine Genauigkeit hinaus wollten die Autoren, dass das Modell wissenschaftliche Einsichten liefert und kein rätselhaftes „Black Box“-System bleibt. Sie verwendeten zwei komplementäre Analysetools, um zu untersuchen, wie stark jeder Eingabefaktor das Modelloutput prägte und wie diese Faktoren miteinander interagierten. Beide Techniken hoben Druck und Molekulargewicht als die wichtigsten Stellgrößen hervor: Höherer Druck erhöhte allgemein die Löslichkeit, weil Kohlendioxid dichter gepackt wird, während schwerere Moleküle tendenziell schlechter löslich waren. Temperatur und Schmelzpunkt spielten subtilere, aber dennoch bedeutende Rollen, hauptsächlich durch Interaktionen mit dem Druck und dadurch, wie fest die Kristallstruktur eines Wirkstoffs zusammenhält. Das Modell reproduzierte auch bekannte thermodynamische Muster, etwa dass eine Temperaturänderung je nach Bereich den Druckeffekt verstärken oder abschwächen kann, was Vertrauen schafft, dass reale physikalische Verhaltensweisen erfasst werden und nicht nur Zahlen auswendig gelernt wurden.
Was die Ergebnisse für die Arzneimittelentwicklung bedeuten
Das leistungsstärkste Hybridmodell, das den Gradient-Boosting-Ansatz mit dem Optimierer inspiriert von gehörnten Eidechsen kombinierte, verringerte den Vorhersagefehler gegenüber einer nicht abgestimmten Baseline um etwa 40 Prozent. Es entsprach nicht nur den mittleren in Experimenten beobachteten Löslichkeitswerten, sondern auch deren Streuung und seltenen Fällen hoher Löslichkeit. Dieses Genauigkeitsniveau erlaubt es dem Modell, als leistungsfähiges Vorscreening-Werkzeug zu fungieren: Forscher können Druck- und Temperaturbereiche auf einem Laptop erkunden, bevor sie sich auf aufwändige Hochdruck-Experimente einlassen, und so Aufwand an schlechten Kandidaten reduzieren. Obwohl der Ansatz noch auf einem überschaubaren Datensatz und einer begrenzten Menge an Wirkstoffeigenschaften beruht und die genaueste Version rechenintensiv ist, zeigt das Framework, wie interpretierbares maschinelles Lernen sowohl die lösungsmittelfreie Formulierung beschleunigen als auch unser Verständnis dafür vertiefen kann, warum manche Wirkstoffe in superkritischem Kohlendioxid besser löslich sind als andere.
Zitation: Khafagy, ES., Lila, A.S.A. & Pishnamazi, M. Accelerating supercritical pharmaceutical formulation via interpretable data-driven prediction of drug solubility. Sci Rep 16, 11006 (2026). https://doi.org/10.1038/s41598-026-44161-9
Schlüsselwörter: Arzneimittel-Löslichkeit, superkritisches Kohlendioxid, maschinelles Lernen, pharmazeutische Formulierung, datengetriebene Modellierung