Clear Sky Science · pt
Huntingtina e seus aliados na sinapse córtico-estriatal
Por que essa proteína cerebral é importante
A doença de Huntington é mais conhecida por causar movimentos bruscos e problemas de memória, mas em sua essência é uma doença de comunicação comprometida entre células cerebrais. Este artigo explora como uma única proteína, a huntingtina, e suas muitas “aliadas” nas junções entre neurônios ajudam a manter os circuitos cerebrais funcionando — e como alterações nessa proteína atrapalham esses circuitos, especialmente ao longo de uma via-chave que conecta pensamento e movimento. Compreender essa fiação oculta oferece ideias novas sobre como retardar ou prevenir a doença.
O centro de tráfego do cérebro para pensamento e movimento
Os autores concentram-se na conexão entre duas regiões cerebrais: a camada externa pensante (córtex) e uma estrutura profunda que ajuda a controlar ações e hábitos (estriado). Neurônios no córtex enviam fibras longas ao estriado, onde formam milhares de pequenos pontos de contato, ou sinapses. Essas sinapses córtico-estriatais estão entre as primeiras estruturas a falhar na doença de Huntington, muito antes que muitas células cerebrais morram. Exames de imagem e estudos em animais mostram que, quando essa via enfraquece, os sintomas pioram. A revisão argumenta que os problemas surgem não apenas nas células estriatais que recebem, como se pensava antes, mas também no lado cortical que envia sinais, que pode na verdade impulsionar grande parte do dano.
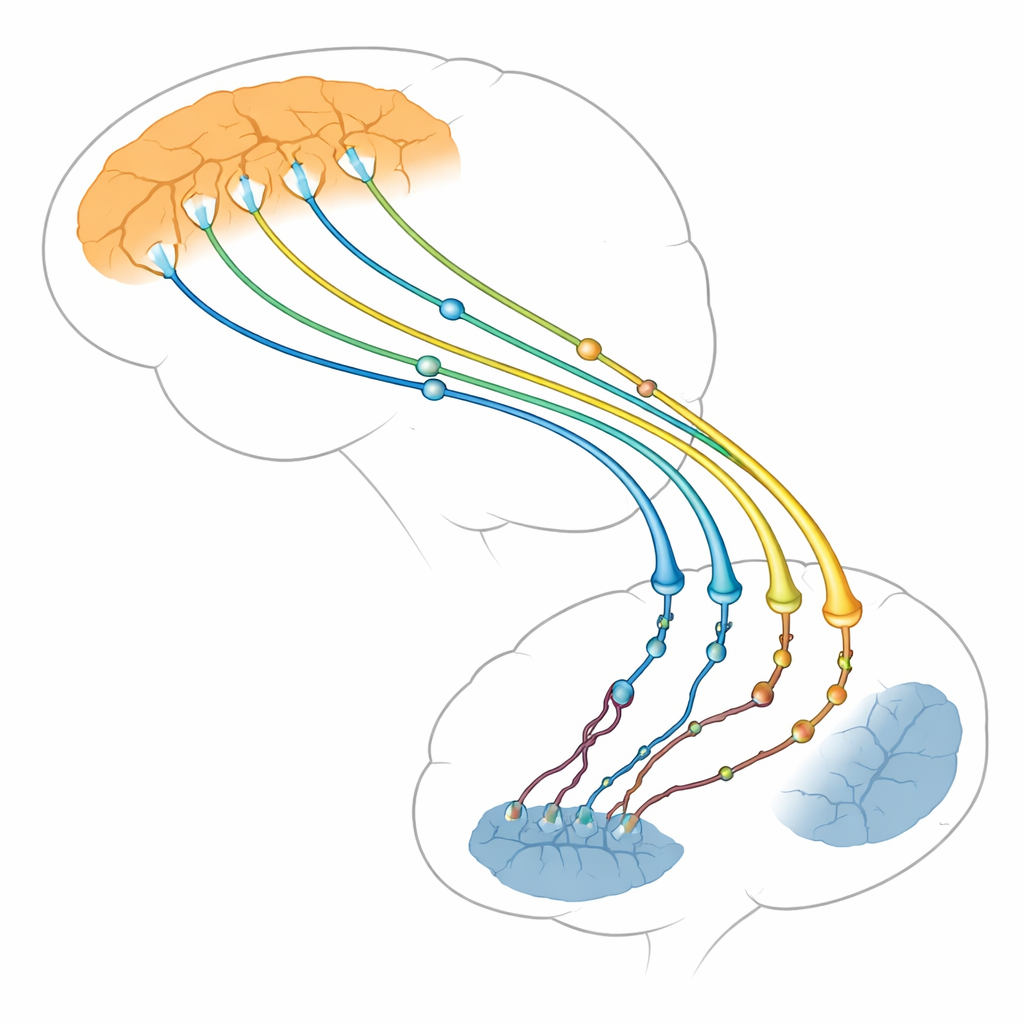
Huntingtina como organizadora mestre das terminações nervosas
A huntingtina é uma proteína grande encontrada por toda a célula nervosa, mas está especialmente concentrada nas sinapses, onde funciona como um andaime ou estação de ancoragem para mais de 3.000 proteínas parceiras. No lado que envia o sinal da sinapse, ela ajuda a mover cargas — como mitocôndrias que produzem energia, vesículas cheias de mensageiros químicos e pacotes de fatores de crescimento chamados BDNF — ao longo de trilhos internos até a terminação nervosa. Também auxilia a posicionar vesículas para liberação, fundi-las com a membrana para expelir seu conteúdo e recuperar a membrana para reciclar novas vesículas. O artigo mostra como a huntingtina, por meio de adaptadores como HAP1 e HIP1 e pequenos interruptores chamados proteínas Rab, coordena esse tráfego constante para que os neurônios possam continuar sinalizando de forma rápida e confiável.
Quando a huntingtina mutante emperra o sistema
Na doença de Huntington, um trecho extra-longo de unidades de glutamina na huntingtina altera sutilmente sua forma e preferências de interação. Essa forma mutante se liga com muita força a alguns parceiros e com pouca força a outros. Como resultado, mitocôndrias fragmentam-se e ficam paradas no corpo celular em vez de alcançar as terminações, pacotes de BDNF viajam mais lentamente e na direção errada, e as vesículas não são adequadamente abastecidas, ancoradas ou recicladas. Proteínas auxiliares chave podem ficar aprisionadas em agregados ricos em huntingtina, e o sistema de limpeza que deveria remover componentes danificados — uma versão especializada da autofagia, ou “auto-comida” celular — funciona com menos eficiência. Com o tempo, o lado que envia sinais da sinapse córtico-estriatal fica privado de energia, suporte de crescimento e vesículas frescas, levando ao desbotamento dos sinais e eventual desconexão.
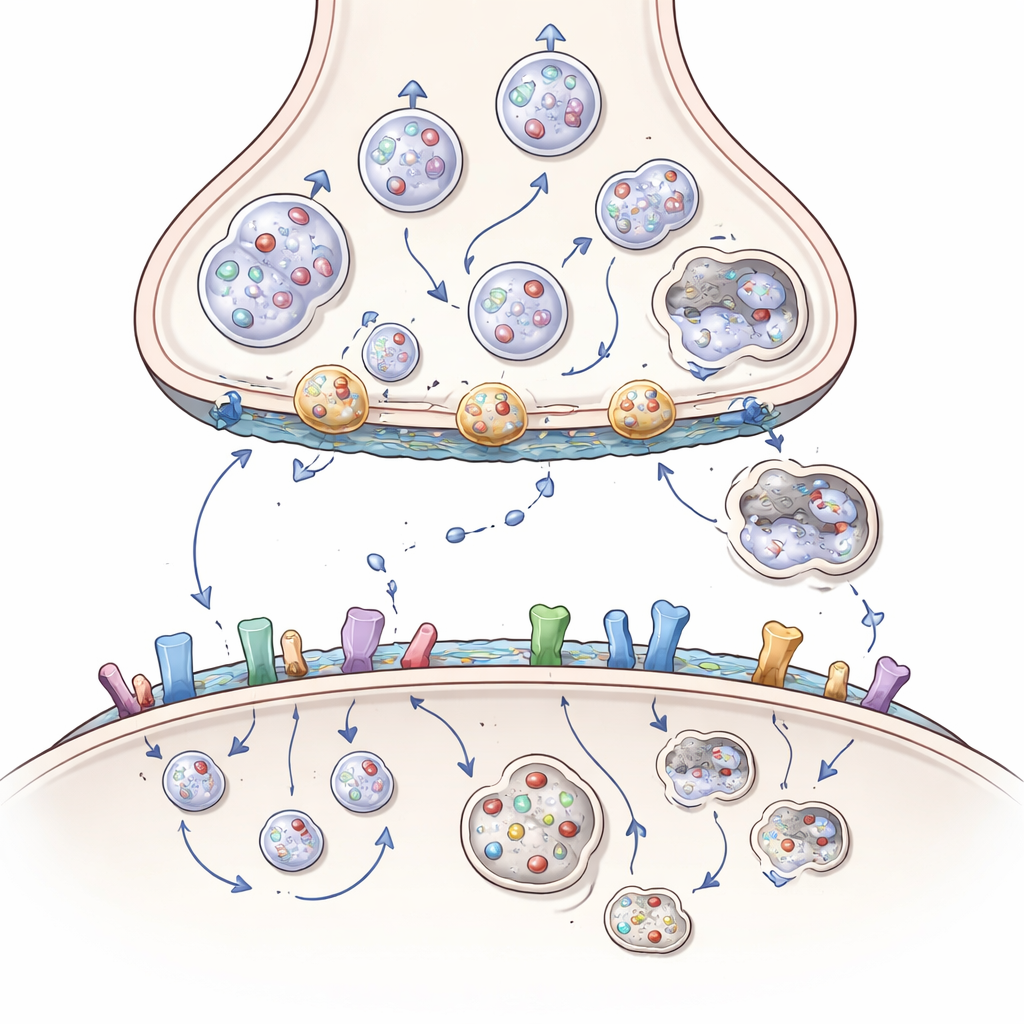
Recepção de sinais, aderência e o papel dos lipídios
O dano não se limita ao lado que envia. No lado que recebe, a huntingtina ajuda a controlar quantos receptores para mensageiros excitatórios e inibitórios ficam na superfície, onde eles se aglomeram e com que rapidez são reciclados. Ao trabalhar com proteínas de andaime e enzimas que acrescentam caudas gordurosas, a huntingtina normalmente mantém certos receptores de glutamato em posições sinápticas protetoras e restringe receptores extrasinápticos mais nocivos. A huntingtina mutante perturba esse equilíbrio, empurrando receptores para locais e combinações que favorecem influxos tóxicos de cálcio e morte celular. Ela também interfere com moléculas de adesão celular que mantêm as sinapses juntas e com lipídios cerebrais, como colesterol e gangliosídeos, que moldam membranas e sustentam a sinalização. A perda desses lipídios na doença de Huntington enfraquece ainda mais as sinapses e a sinalização por fatores de crescimento.
Novos ângulos para terapia
A revisão conclui que a huntingtina não é apenas uma vítima passiva da mutação, mas uma organizadora ativa das sinapses cujas parcerias se desordenam na doença. Como muitos de seus aliados mais críticos ficam no lado pré-sináptico de neurônios corticais vulneráveis, visar esses parceiros — como adaptadores de transporte, carregadores de fatores de crescimento, enzimas chave como ADAM10 ou vias lipídicas — pode oferecer novas rotas para proteger as sinapses córtico-estriatais sem a necessidade de eliminar a huntingtina por completo. Avanços futuros em imagem de super-resolução, modelos “cérebro-em-um-chip” e o estudo das modificações químicas da huntingtina podem revelar precisamente onde e quando intervir, abrindo esperanças para terapias que preservem as linhas de comunicação do cérebro e retardem a progressão da Huntington.
Citação: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Palavras-chave: Doença de Huntington, sinapse córtico-estriatal, proteína huntingtina, disfunção sináptica, transporte axonal