Clear Sky Science · es
Huntingtina y sus aliados en la sinapsis córtico-estriatal
Por qué importa esta proteína cerebral
La enfermedad de Huntington es más conocida por provocar movimientos bruscos y problemas de memoria, pero en esencia es una enfermedad de comunicación rota entre las neuronas. Este artículo explora cómo una única proteína, la huntingtina, y sus numerosos “aliados” en las uniones entre células nerviosas ayudan a mantener los circuitos cerebrales en funcionamiento —y cómo las alteraciones en esta proteína descarrilan esos circuitos, especialmente a lo largo de una vía clave que conecta el pensamiento con el movimiento. Comprender este cableado oculto ofrece ideas nuevas sobre cómo frenar o prevenir la enfermedad.
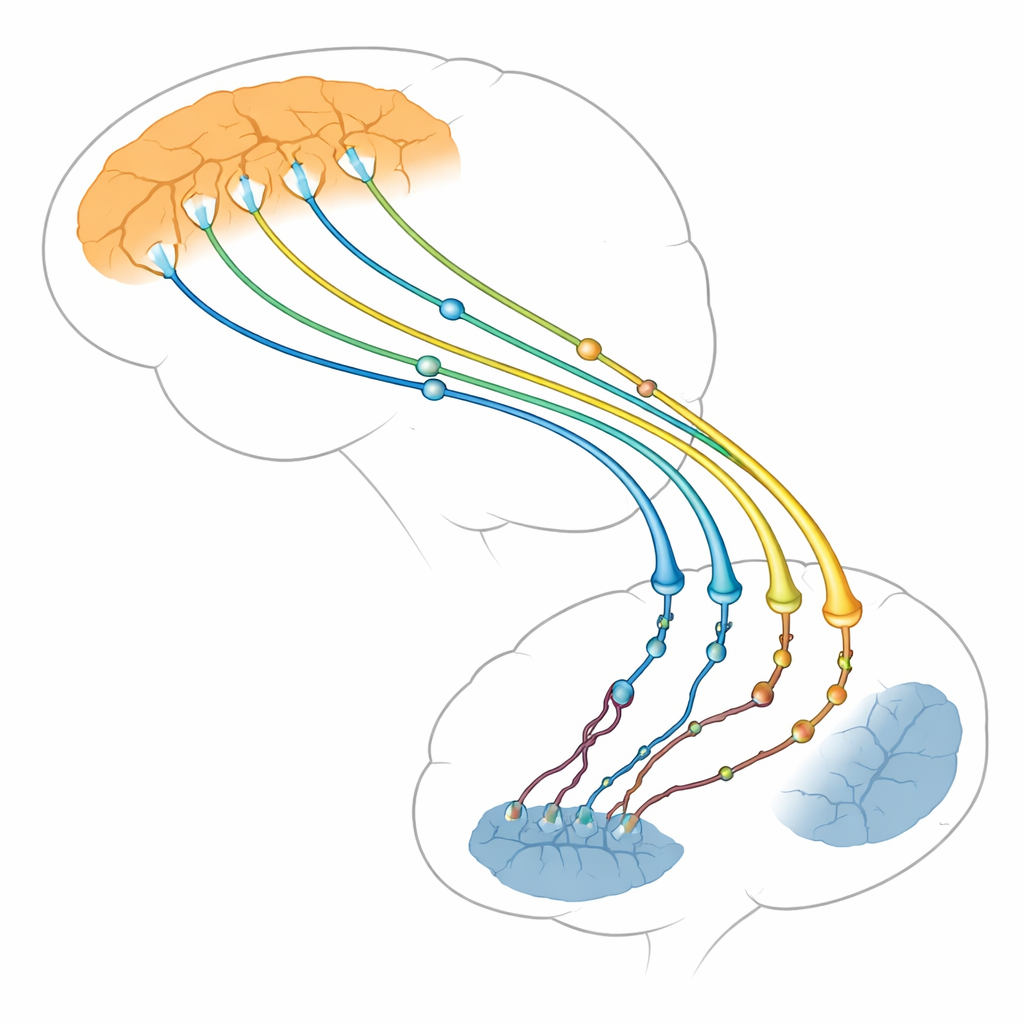
El centro de tráfico cerebral para pensamiento y movimiento
Los autores se centran en la conexión entre dos regiones cerebrales: la capa externa pensante (corteza) y una estructura profunda que ayuda a controlar acciones y hábitos (estriado). Las neuronas de la corteza envían fibras largas hacia el estriado, donde forman miles de puntos de contacto diminutos, o sinapsis. Estas sinapsis córtico-estriatales están entre las primeras estructuras en fallar en la enfermedad de Huntington, mucho antes de que mueran muchas neuronas. Imágenes cerebrales y estudios en animales muestran que cuando esta vía se debilita, los síntomas empeoran. La revisión sostiene que los problemas no surgen solo en las células receptoras del estriado, como se pensaba antes, sino también en el lado cortical emisor, que puede ser el impulsor principal del daño.
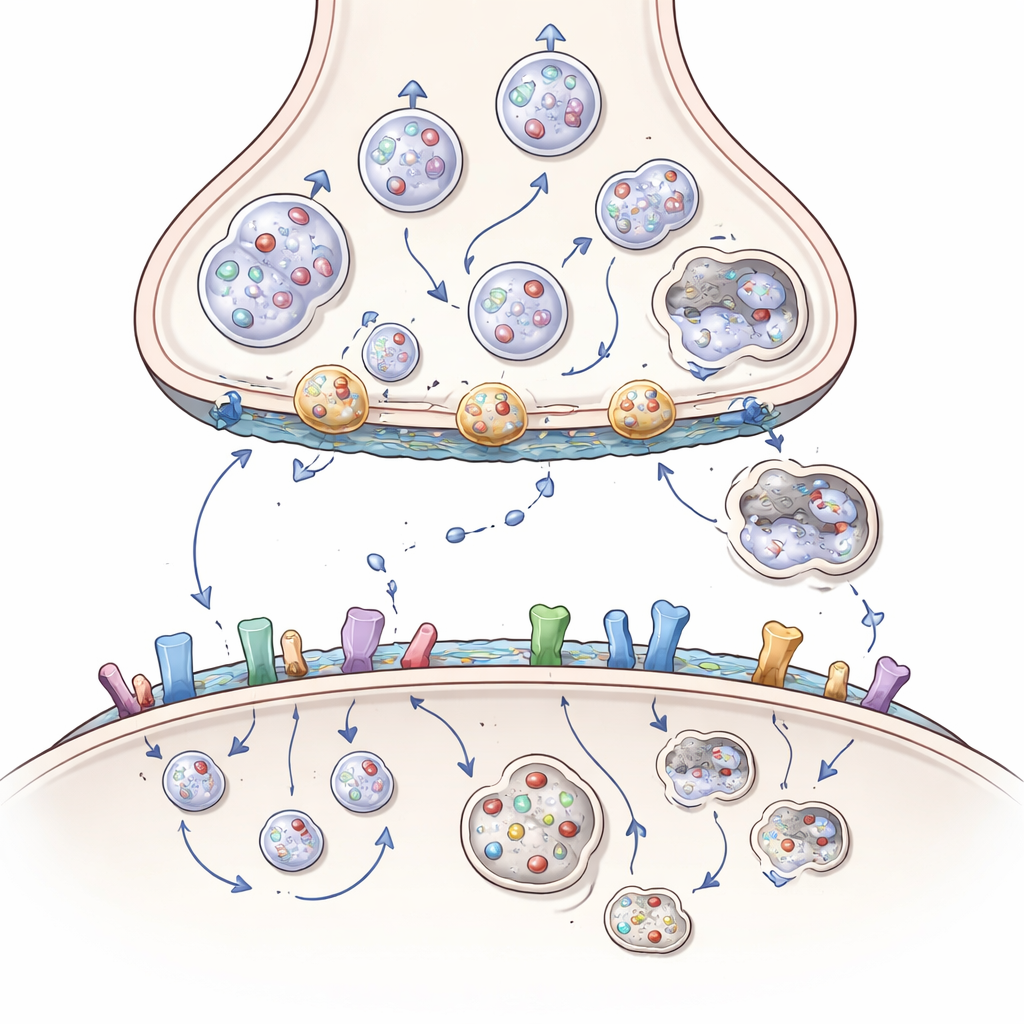
La huntingtina como organizadora maestra de las terminaciones nerviosas
La huntingtina es una proteína grande presente en toda la neurona, pero está especialmente concentrada en las sinapsis, donde actúa como andamiaje o estación de acoplamiento para más de 3.000 proteínas asociadas. En el lado emisor de la sinapsis, ayuda a mover carga —como mitocondrias productoras de energía, vesículas llenas de mensajeros químicos y paquetes de factor de crecimiento llamados BDNF— a lo largo de pistas internas hasta la terminación nerviosa. También contribuye a situar las vesículas para su liberación, fusionarlas con la membrana para descargar su contenido y recuperar la membrana para reciclar nuevas vesículas. El artículo muestra cómo la huntingtina, a través de adaptadores como HAP1 y HIP1 y pequeños interruptores llamados proteínas Rab, coordina ese tráfico constante para que las neuronas puedan transmitir señales de forma rápida y fiable.
Cuando la huntingtina mutante atasca el sistema
En la enfermedad de Huntington, una cadena extra larga de unidades de glutamina en la huntingtina cambia sutilmente su forma y sus preferencias de unión. Esta forma mutante se aferra con demasiada fuerza a algunos socios y con demasiado poca a otros. Como resultado, las mitocondrias se fragmentan y se quedan detenidas en el cuerpo celular en lugar de alcanzar las terminaciones nerviosas, los paquetes de BDNF viajan más despacio y en la dirección equivocada, y las vesículas no se aprovisionan, anclan ni reciclan correctamente. Proteínas auxiliares clave pueden quedar atrapadas en agregados ricos en huntingtina, y el sistema de limpieza que debería eliminar componentes dañados —una versión especializada de la autofagia celular— funciona con menos eficacia. Con el tiempo, el lado emisor de la sinapsis córtico-estriatal se queda sin energía, sin apoyo de factores de crecimiento y sin vesículas nuevas, lo que provoca señales débiles y, eventualmente, la desconexión.
Recepción de la señal, adhesividad y el papel de las grasas
El daño no se limita al lado emisor. En el lado receptor, la huntingtina ayuda a controlar cuántos receptores para mensajeros excitadores e inhibidores se sitúan en la superficie, dónde se agrupan y con qué rapidez se reciclan. Al asociarse con proteínas andamiaje y enzimas que añaden colas grasas, la huntingtina normalmente mantiene ciertos receptores de glutamato en posiciones sinápticas protectoras y restringe receptores extrasinápticos más perjudiciales. La huntingtina mutante altera este equilibrio, empujando receptores hacia ubicaciones y combinaciones que favorecen una entrada tóxica de calcio y la muerte celular. También interfiere con moléculas de adhesión celular que mantienen las sinapsis unidas y con lípidos cerebrales como el colesterol y las gangliósidos que configuran las membranas y apoyan la señalización. La pérdida de estos lípidos en la enfermedad de Huntington debilita aún más las sinapsis y la señalización de factores de crecimiento.
Nuevas vías para la terapia
La revisión concluye que la huntingtina no es solo una víctima pasiva de la mutación, sino una organizadora activa de las sinapsis cuyas asociaciones se trastocan en la enfermedad. Dado que muchos de sus aliados más críticos están en el lado presináptico de las neuronas corticales vulnerables, dirigir a estos socios —como adaptadores de transporte, transportadores de factores de crecimiento, enzimas clave como ADAM10 o vías lipídicas— podría ofrecer nuevas rutas para proteger las sinapsis córtico-estriatales sin necesidad de eliminar la huntingtina por completo. Avances futuros en imagen superresolutiva, modelos “cerebro-en-un-chip” y el estudio de las modificaciones químicas de la huntingtina podrían revelar con precisión dónde y cuándo intervenir, lo que aumenta la esperanza de terapias que preserven las líneas de comunicación del cerebro y ralenticen la progresión de la Huntington.
Cita: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Palabras clave: Enfermedad de Huntington, sinapsis córtico-estriatal, proteína huntingtina, disfunción sináptica, transporte axonal