Clear Sky Science · pl
Huntingtyna i jej sojusznicy w synapsie korowo‑prążkowiowej
Dlaczego to białko mózgu ma znaczenie
Choroba Huntingtona jest najbardziej znana z wywoływania szarpanych ruchów i problemów z pamięcią, ale w istocie to choroba zaburzonej komunikacji między komórkami mózgu. Artykuł omawia, jak jedno białko — huntingtyna — i jego liczni „sojusznicy” w miejscach styku neuronów pomagają utrzymać funkcjonowanie obwodów mózgowych — oraz jak zmiany w tym białku wykolejają te obwody, szczególnie wzdłuż kluczowej trasy łączącej myślenie z ruchem. Zrozumienie tej ukrytej sieci dostarcza świeżych pomysłów na spowolnienie lub zapobieganie chorobie.
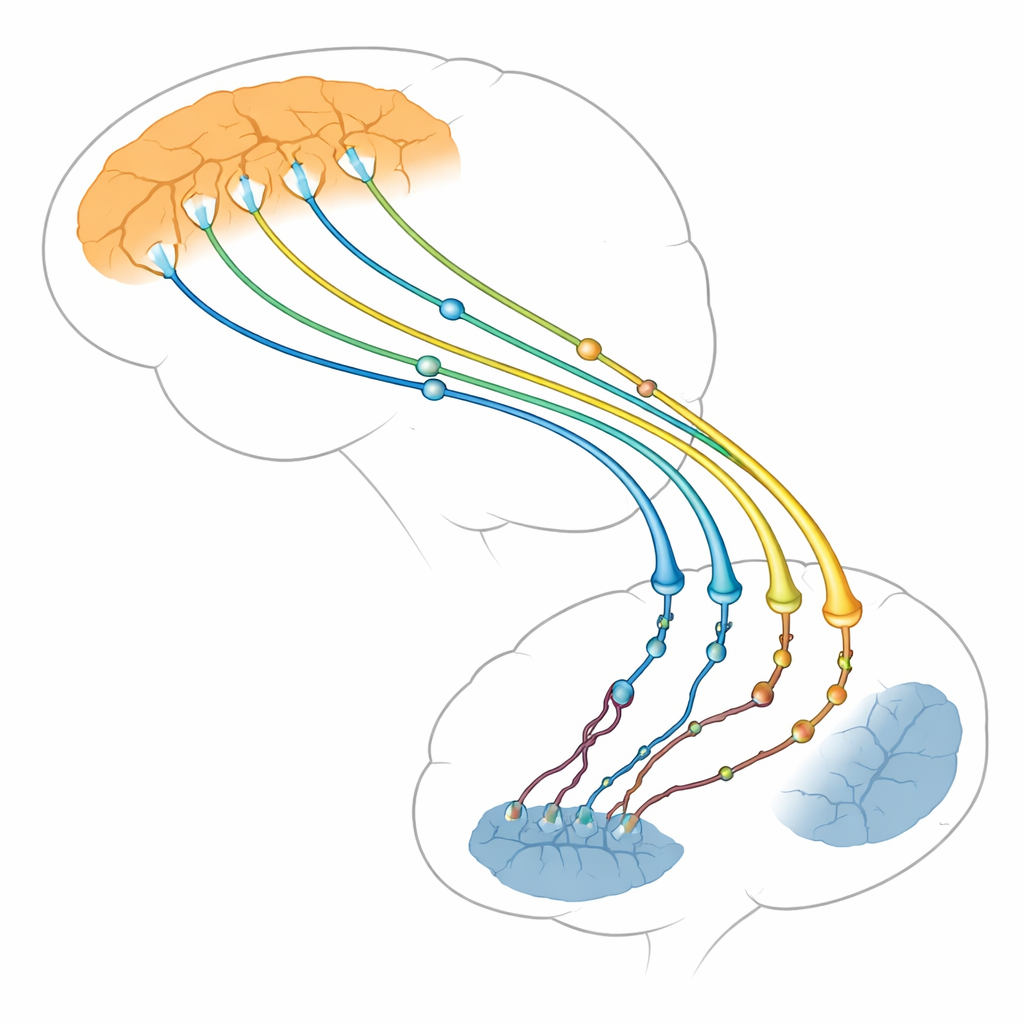
Węzeł komunikacyjny mózgu dla myśli i ruchu
Autorzy koncentrują się na połączeniu między dwiema strukturami mózgu: myślącą, zewnętrzną warstwą (kora) a głęboką strukturą, która pomaga kontrolować działania i nawyki (prążkowie). Komórki nerwowe w korze wysyłają długie włókna do prążkowia, gdzie tworzą tysiące drobnych punktów kontaktowych, czyli synaps. Synapsy korowo‑prążkowiowe należą do pierwszych struktur, które zawodzą w chorobie Huntingtona, na długo przed obumieraniem wielu komórek mózgowych. Badania obrazowe mózgu i modele zwierzęce pokazują, że gdy ta droga słabnie, objawy się nasilają. Przegląd argumentuje, że problemy pojawiają się nie tylko po stronie odbierającej w prążkowiu, jak sądzono dawniej, lecz również po stronie wysyłającej w korze, która może w rzeczywistości napędzać dużą część uszkodzeń.
Huntingtyna jako główny organizator zakończeń nerwowych
Huntingtyna to duże białko występujące w całej komórce nerwowej, ale szczególnie bogate w synapsach, gdzie działa jak szkielet lub stacja dokująca dla ponad 3 000 białkowych partnerów. Po stronie wysyłającej synapsy pomaga przemieszczać ładunki — takie jak mitochondria produkujące energię, pęcherzyki wypełnione neuroprzekaźnikami oraz pakiety czynników wzrostu, jak BDNF — wzdłuż wewnętrznych torów do zakończenia nerwowego. Pomaga też ustawiać pęcherzyki do uwolnienia, łączyć je z błoną by uwalniały zawartość oraz odzyskiwać błonę, by zrecyklować nowe pęcherzyki. Artykuł pokazuje, jak huntingtyna, poprzez adaptorów takich jak HAP1 i HIP1 oraz małe przełączniki zwane białkami Rab, koordynuje ten nieustanny ruch, by neurony mogły wysyłać sygnały szybko i niezawodnie.
Gdy zmutowana huntingtyna zatyka mechanizmy
W chorobie Huntingtona nadmiernie wydłużony odcinek jednostek glutaminy w huntingtynie subtelnie zmienia jej kształt i preferencje wiązania. Zmutowana forma przywiera zbyt mocno do niektórych partnerów, a zbyt słabo do innych. W efekcie mitochondria fragmentują się i zatrzymują w ciele komórki zamiast docierać do zakończeń, pakiety BDNF poruszają się wolniej i w złym kierunku, a pęcherzyki nie są właściwie zaopatrzone, zadokowane ani recyklingowane. Kluczowe białka pomocnicze mogą zostać uwięzione w skupiskach bogatych w huntingtynę, a system sprzątający, który powinien usuwać uszkodzone komponenty — wyspecjalizowana forma autophagii — działa mniej efektywnie. Z czasem strona wysyłająca synapsy korowo‑prążkowiowej zostaje pozbawiona energii, wsparcia wzrostowego i świeżych pęcherzyków, co prowadzi do gasnących sygnałów i w końcu do rozłączenia.
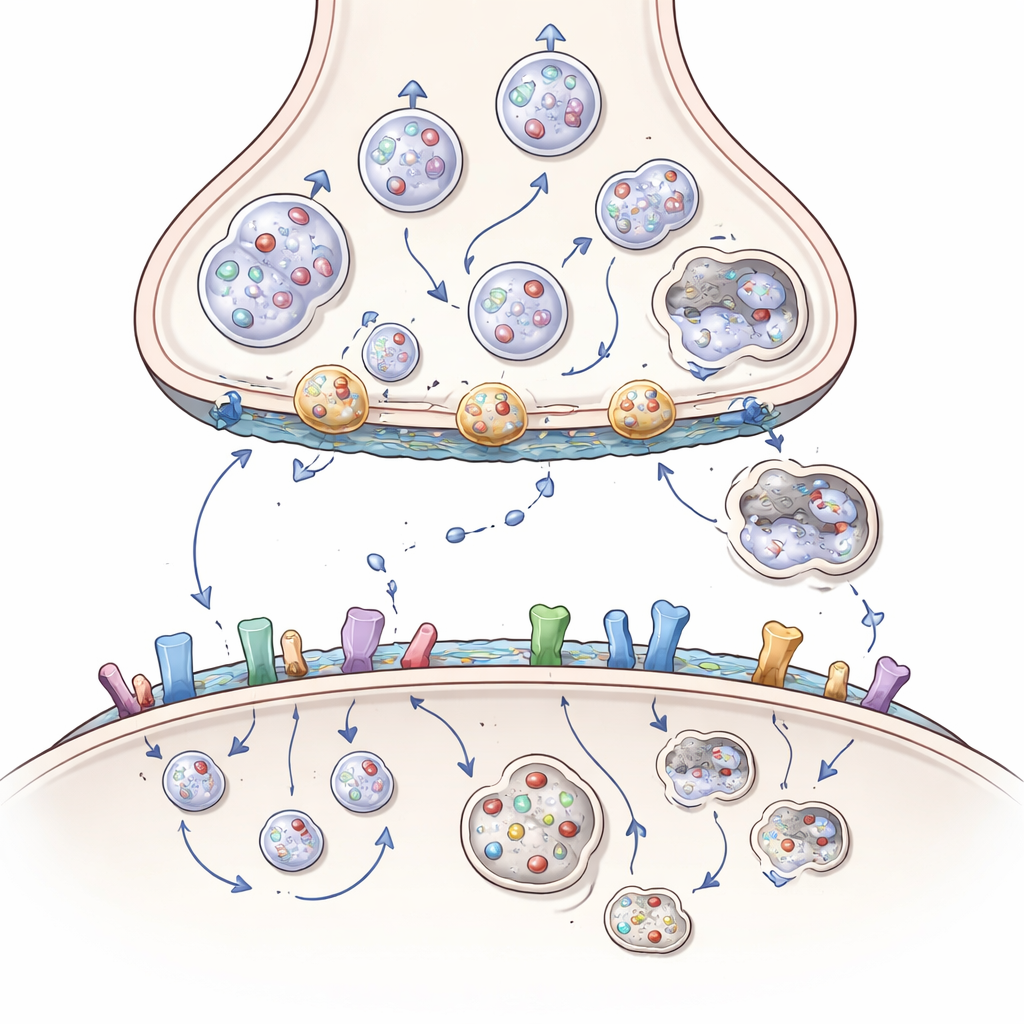
Odbiór sygnału, przylepność i rola tłuszczów
Uszkodzenia nie ograniczają się do strony wysyłającej. Po stronie odbierającej huntingtyna pomaga kontrolować, ile receptorów dla pobudzających i hamujących przekaźników znajduje się na powierzchni, jak się grupują i jak szybko są recyklowane. Współdziałając ze białkami rusztowania i enzymami przyłączającymi tłuszczowe łańcuchy, huntingtyna zwykle utrzymuje niektóre receptory glutaminianowe w ochronnych pozycjach synaptycznych i powstrzymuje bardziej szkodliwe, pozasynaptyczne warianty. Zmutowana huntingtyna zaburza tę równowagę, przesuwając receptory w miejsca i konfiguracje sprzyjające toksycznemu napływowi wapnia i śmierci komórki. Zakłóca też cząsteczki adhezji komórkowej utrzymujące synapsy oraz lipidy mózgowe, takie jak cholesterol i gangliozydy, które kształtują błony i wspierają przekazywanie sygnałów. Utrata tych lipidów w chorobie Huntingtona dodatkowo osłabia synapsy i sygnalizację czynników wzrostu.
Nowe kąty terapeutyczne
Przegląd konkluduje, że huntingtyna nie jest jedynie bierną ofiarą mutacji, lecz aktywnym organizatorem synaps, którego partnerstwa w chorobie idą w złym kierunku. Ponieważ wielu jej najważniejszych sojuszników znajduje się po presynaptycznej stronie podatnych neuronów korowych, ukierunkowanie na tych partnerów — takich jak adaptory transportowe, nośniki czynników wzrostu, kluczowe enzymy typu ADAM10 czy szlaki lipidowe — może dać nowe możliwości ochrony synaps korowo‑prążkowiowych bez konieczności całkowitego eliminowania huntingtyny. Przyszłe postępy w obrazowaniu o superrozdzielczości, modelach „mózg‑na‑chipie” i badaniu chemicznych modyfikacji huntingtyny mogą ujawnić dokładnie gdzie i kiedy interweniować, dając nadzieję na terapie, które zachowają linie komunikacyjne mózgu i spowolnią postęp choroby Huntingtona.
Cytowanie: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Słowa kluczowe: choroba Huntingtona, synapsa korowo‑prążkowiowa, białko huntingtyna, dysfunkcja synaptyczna, transport aksonalny