Clear Sky Science · en
Huntingtin and its allies at the cortico-striatal synapse
Why this brain protein matters
Huntington’s disease is best known for causing jerky movements and memory problems, but at its core it is a disease of broken communication between brain cells. This article explores how a single protein, huntingtin, and its many “allies” at the junctions between nerve cells help keep brain circuits working—and how changes in this protein derail those circuits, especially along a key highway linking thought and movement. Understanding this hidden wiring offers fresh ideas for how to slow or prevent the disease.
The brain’s traffic hub for thought and movement
The authors focus on the connection between two brain regions: the thinking outer layer (cortex) and a deep structure that helps control actions and habits (striatum). Nerve cells in the cortex send long fibers down to the striatum, where they form thousands of tiny contact points, or synapses. These cortico-striatal synapses are among the first structures to falter in Huntington’s disease, long before many brain cells die. Brain scans and animal studies show that when this pathway weakens, symptoms worsen. The review argues that problems arise not just in the receiving striatal cells, as once thought, but also in the sending cortical side, which may actually drive much of the damage.
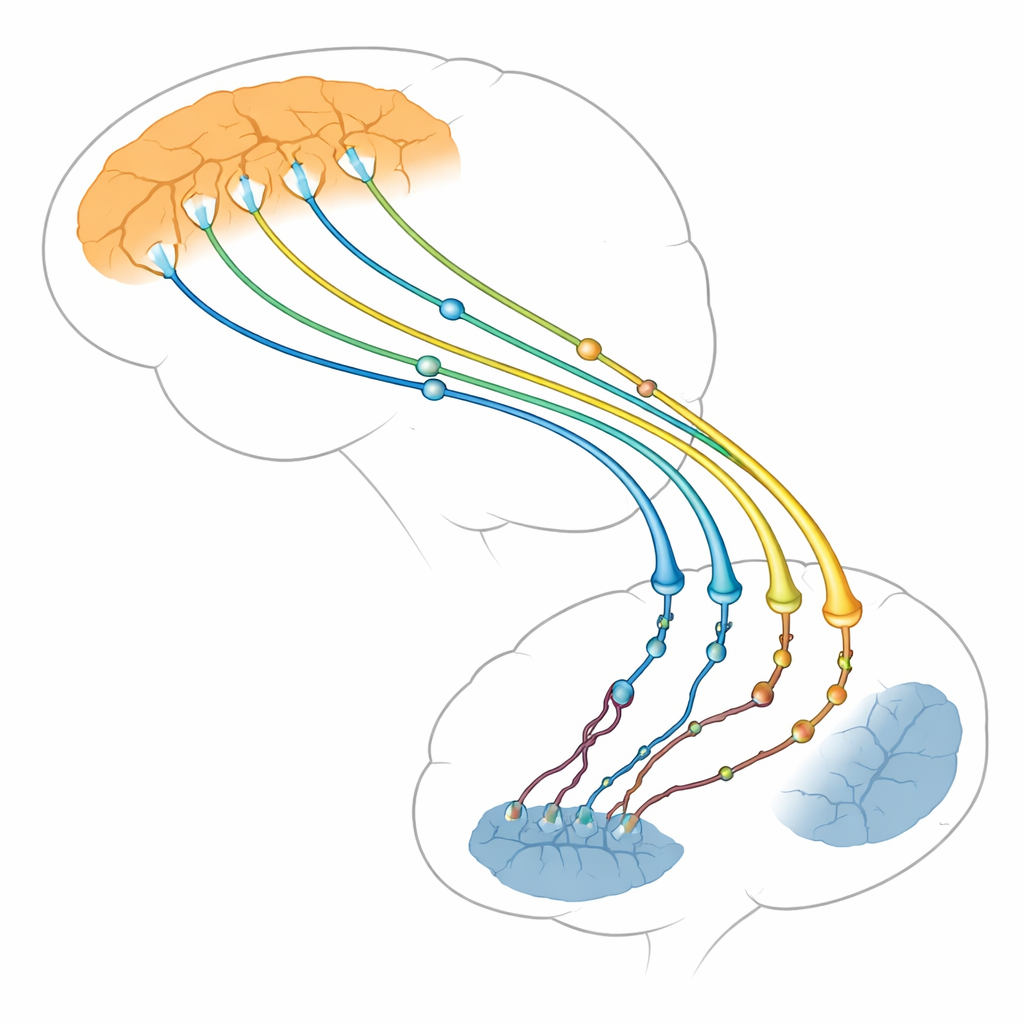
Huntingtin as a master organizer of nerve endings
Huntingtin is a large protein found throughout nerve cells, but it is especially enriched at synapses, where it acts like a scaffold or docking station for more than 3,000 partner proteins. At the sending side of the synapse, it helps move cargo—such as energy-producing mitochondria, chemical-filled vesicles, and growth-factor packets called BDNF—along internal tracks to the nerve ending. It also helps position vesicles for release, fuse them with the membrane to discharge their contents, and pull the membrane back in to recycle new vesicles. The article shows how huntingtin, through adaptors like HAP1 and HIP1 and small switches called Rab proteins, coordinates this constant traffic so that nerve cells can keep signaling rapidly and reliably.
When mutant huntingtin jams the works
In Huntington’s disease, an extra-long stretch of glutamine units in huntingtin subtly changes its shape and binding preferences. This mutant form clings too tightly to some partners and too weakly to others. As a result, mitochondria fragment and stall in the cell body instead of reaching nerve endings, BDNF packets travel more slowly and in the wrong direction, and vesicles are not properly stocked, docked, or recycled. Key helper proteins can become trapped in huntingtin-rich clumps, and the clean-up system that should remove damaged components—a specialized version of cellular self-eating called autophagy—works less efficiently. Over time, the sending side of the cortico-striatal synapse becomes starved of energy, growth support, and fresh vesicles, leading to fading signals and eventual disconnection.
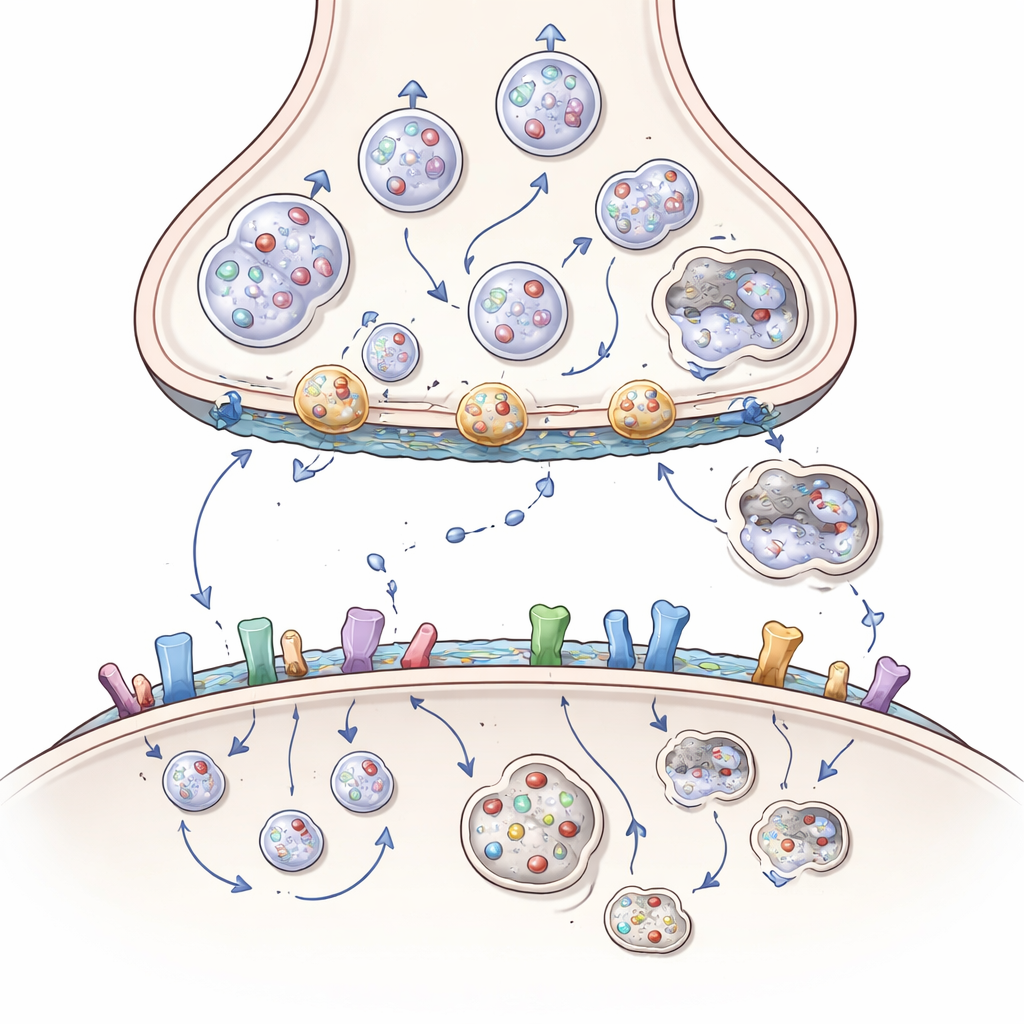
Signal reception, stickiness, and the role of fats
The damage is not limited to the sending side. On the receiving side, huntingtin helps control how many receptors for excitatory and inhibitory messengers sit on the surface, where they cluster, and how quickly they are recycled. By partnering with scaffold proteins and enzymes that attach fatty tails, huntingtin normally keeps certain glutamate receptors in protective synaptic positions and restrains more harmful, extrasynaptic ones. Mutant huntingtin upsets this balance, pushing receptors toward locations and combinations that favor toxic calcium influx and cell death. It also interferes with cell-adhesion molecules that hold synapses together and with brain lipids such as cholesterol and gangliosides that shape membranes and support signaling. The loss of these lipids in Huntington’s disease further weakens synapses and growth-factor signaling.
New angles for therapy
The review concludes that huntingtin is not just a passive victim of mutation but an active organizer of synapses whose partnerships go awry in disease. Because many of its most critical allies sit on the presynaptic side of vulnerable cortical neurons, targeting these partners—such as transport adaptors, growth-factor carriers, key enzymes like ADAM10, or lipid pathways—could offer new routes to protect cortico-striatal synapses without needing to eliminate huntingtin entirely. Future advances in super-resolution imaging, “brain-on-a-chip” models, and the study of huntingtin’s chemical modifications may reveal precisely where and when to intervene, raising hope for therapies that preserve the brain’s communication lines and slow Huntington’s progression.
Citation: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Keywords: Huntington’s disease, cortico-striatal synapse, huntingtin protein, synaptic dysfunction, axonal transport