Clear Sky Science · it
Huntingtin e i suoi alleati alla sinapsi cortico-striatale
Perché questa proteina cerebrale importa
La malattia di Huntington è soprattutto nota per provocare movimenti scoordinati e problemi di memoria, ma alla base è una malattia della comunicazione interrotta fra cellule cerebrali. Questo articolo esplora come una singola proteina, la huntingtin, e i suoi numerosi “alleati” alle giunzioni tra i neuroni contribuiscano a mantenere in funzione i circuiti cerebrali — e come le alterazioni di questa proteina mandino fuori strada quei circuiti, in particolare lungo un’autostrada chiave che mette in collegamento pensiero e movimento. Comprendere questo cablaggio nascosto offre nuove idee su come rallentare o prevenire la malattia.
Il nodo del traffico cerebrale per pensiero e movimento
Gli autori si concentrano sulla connessione tra due aree cerebrali: lo strato esterno pensante (corteccia) e una struttura profonda che aiuta a controllare azioni e abitudini (striatum). I neuroni corticali inviano lunghi prolungamenti verso lo striato, dove formano migliaia di minuscoli punti di contatto, o sinapsi. Queste sinapsi cortico-striatali sono tra le prime strutture a vacillare nella malattia di Huntington, molto prima che molte cellule cerebrali muoiano. Risonanze cerebrali e studi su animali mostrano che quando questo percorso si indebolisce, i sintomi peggiorano. La rassegna sostiene che i problemi non nascono solo nei neuroni striatali riceventi, come si pensava un tempo, ma anche nel lato corticale mittente, che potrebbe in realtà guidare gran parte del danno.
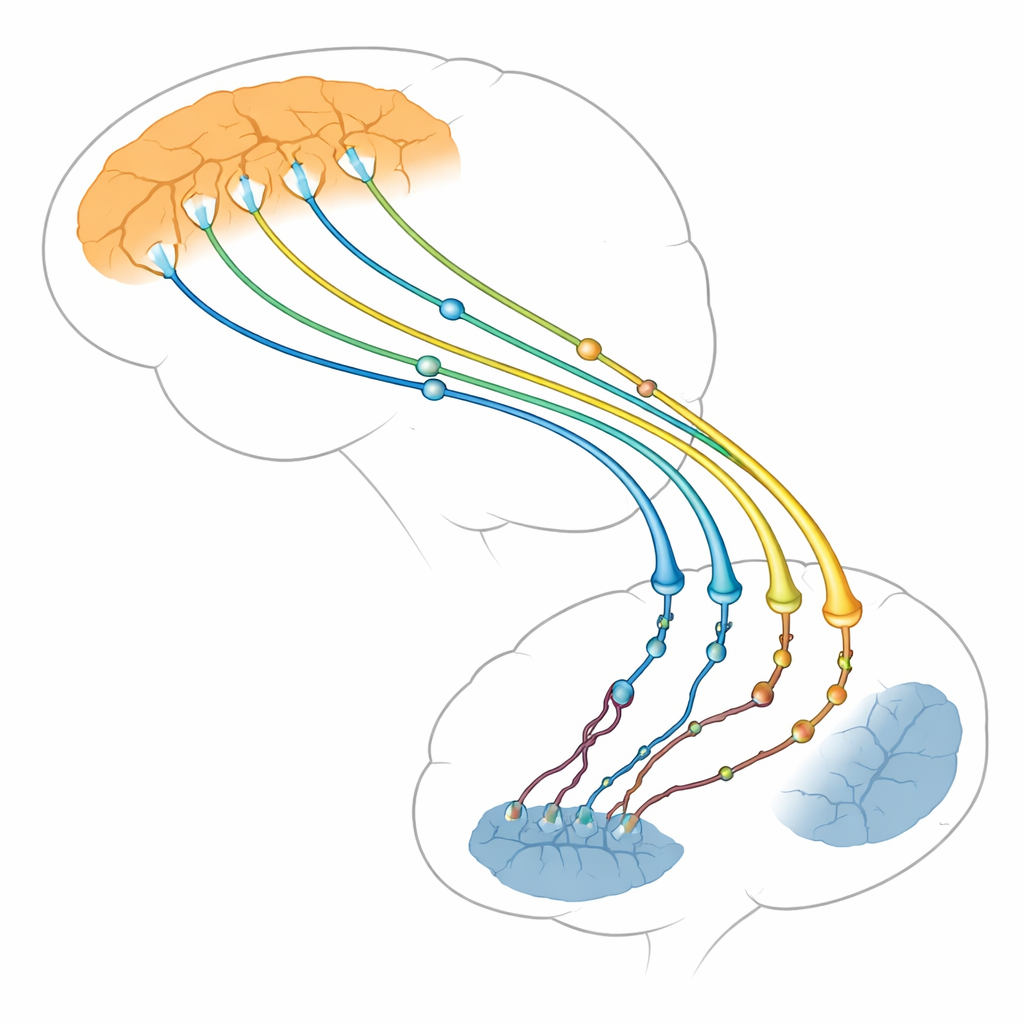
La huntingtin come organizzatrice principale delle terminazioni nervose
La huntingtin è una proteina di grandi dimensioni presente in tutto il neurone, ma è particolarmente concentrata alle sinapsi, dove funge da impalcatura o piattaforma di aggancio per oltre 3.000 proteine partner. Sul lato mittente della sinapsi aiuta a spostare il carico — come mitocondri che producono energia, vescicole piene di neurotrasmettitori e pacchetti di fattori di crescita come il BDNF — lungo i binari interni fino alla terminazione nervosa. Aiuta anche a posizionare le vescicole per il rilascio, a fonderle con la membrana per scaricarne il contenuto e a recuperare la membrana per riciclare nuove vescicole. L’articolo mostra come la huntingtin, tramite adattatori come HAP1 e HIP1 e piccoli interruttori detti proteine Rab, coordini questo traffico costante in modo che i neuroni possano continuare a segnalare rapidamente e in modo affidabile.
Quando la huntingtin mutata inceppa il sistema
Nella malattia di Huntington, una striscia troppo lunga di unità glutammato nella huntingtin ne modifica sottilmente la forma e le preferenze di legame. Questa forma mutante si lega troppo saldamente ad alcuni partner e troppo debolmente ad altri. Di conseguenza, i mitocondri si frammentano e si bloccano nel corpo cellulare invece di raggiungere le terminazioni nervose, i pacchetti di BDNF viaggiano più lentamente e nella direzione sbagliata, e le vescicole non sono adeguatamente caricate, ancorate o riciclate. Proteine helper chiave possono rimanere intrappolate in ammassi ricchi di huntingtin, e il sistema di pulizia che dovrebbe rimuovere componenti danneggiati — una versione specializzata dell’autofagia, un processo di auto-digestione cellulare — funziona con meno efficienza. Col tempo, il lato mittente della sinapsi cortico-striatale rimane privo di energia, supporto per la crescita e vescicole fresche, portando al deterioramento dei segnali e alla disconnessione finale.
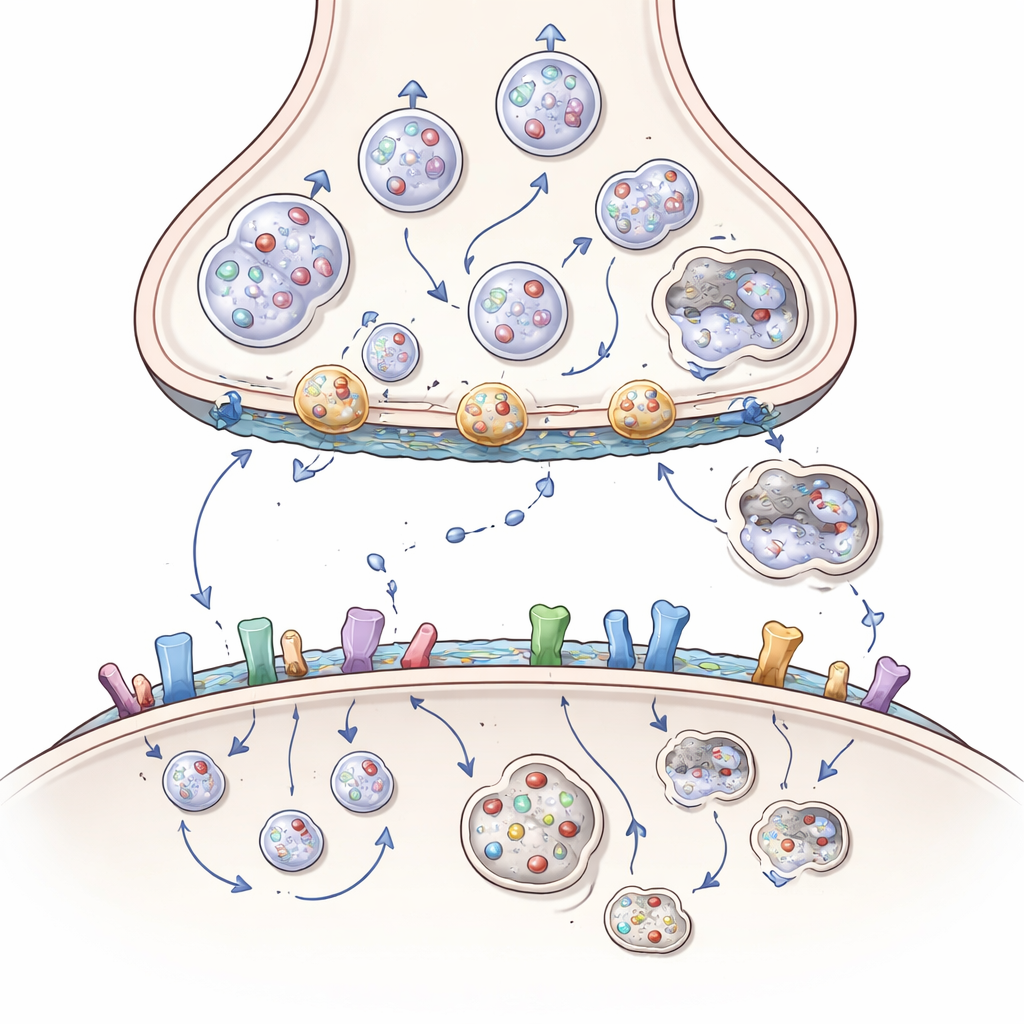
Ricezione del segnale, adesività e il ruolo dei lipidi
Il danno non si limita al lato mittente. Sul lato ricevente, la huntingtin aiuta a controllare quante proteine recettrici per messaggeri eccitatori e inibitori sono presenti sulla superficie, come si raggruppano e quanto rapidamente vengono riciclate. Collaborando con proteine impalcatura e enzimi che attaccano code lipidiche, la huntingtin normalmente mantiene alcuni recettori del glutammato in posizioni sinaptiche protettive e frena quelli più dannosi extrasinaptici. La huntingtin mutata altera questo equilibrio, spingendo i recettori verso localizzazioni e combinazioni che favoriscono un influx nocivo di calcio e la morte cellulare. Interferisce anche con molecole di adesione cellulare che tengono insieme le sinapsi e con lipidi cerebrali come colesterolo e gangliosidi che modellano le membrane e supportano la segnalazione. La perdita di questi lipidi nella malattia di Huntington indebolisce ulteriormente sinapsi e la trasmissione dei fattori di crescita.
Nuove piste per la terapia
La rassegna conclude che la huntingtin non è solo una vittima passiva della mutazione ma un’organizzatrice attiva delle sinapsi le cui partnership si guastano nella malattia. Poiché molti dei suoi alleati più critici si trovano sul lato presinaptico dei neuroni corticali vulnerabili, prendere di mira questi partner — come adattatori del trasporto, vettori di fattori di crescita, enzimi chiave come ADAM10 o vie lipidiche — potrebbe offrire nuove strade per proteggere le sinapsi cortico-striatali senza dover eliminare completamente la huntingtin. Progressi futuri in imaging super-risolutivo, modelli “brain-on-a-chip” e nello studio delle modificazioni chimiche della huntingtin potrebbero rivelare esattamente dove e quando intervenire, alimentando la speranza per terapie che preservino le linee di comunicazione cerebrale e rallentino la progressione della malattia di Huntington.
Citazione: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Parole chiave: Malattia di Huntington, sinapsi cortico-striatale, proteina huntingtin, disfunzione sinaptica, trasporto assonale