Clear Sky Science · de
Huntingtin und seine Partner an der kortiko-striatalen Synapse
Warum dieses Gehirnprotein wichtig ist
Die Huntington-Krankheit ist vor allem für ruckartige Bewegungen und Gedächtnisprobleme bekannt, doch im Kern handelt es sich um eine Erkrankung der gestörten Kommunikation zwischen Nervenzellen. Dieser Artikel untersucht, wie ein einzelnes Protein, Huntingtin, und seine zahlreichen „Partner“ an den Verbindungsstellen zwischen Nervenzellen dazu beitragen, Gehirnnetzwerke funktionsfähig zu halten – und wie Veränderungen dieses Proteins diese Netzwerke entgleisen lassen, insbesondere entlang einer zentralen Verkehrsader, die Denken und Bewegung verbindet. Das Verständnis dieser verborgenen Verdrahtung liefert neue Ansätze, um das Fortschreiten der Erkrankung zu verlangsamen oder zu verhindern.
Das Verkehrszentrum des Gehirns für Denken und Bewegung
Die Autoren konzentrieren sich auf die Verbindung zwischen zwei Hirnregionen: der denkenden Außenschicht (Kortex) und einer tiefen Struktur, die bei der Steuerung von Handlungen und Gewohnheiten hilft (Striatum). Neurone im Kortex senden lange Fasern zum Striatum, wo sie Tausende winziger Kontaktpunkte, so genannter Synapsen, bilden. Diese kortiko-striatalen Synapsen gehören zu den ersten Strukturen, die bei der Huntington-Krankheit versagen, lange bevor viele Nervenzellen absterben. Bildgebende Untersuchungen und Tierstudien zeigen, dass mit der Schwächung dieser Bahn die Symptome zunehmen. Die Übersichtsarbeit argumentiert, dass die Probleme nicht nur in den empfangenden striatalen Zellen auftreten, wie früher angenommen, sondern auch auf der sendenden kortikalen Seite, die tatsächlich einen großen Teil des Schadens antreiben könnte.
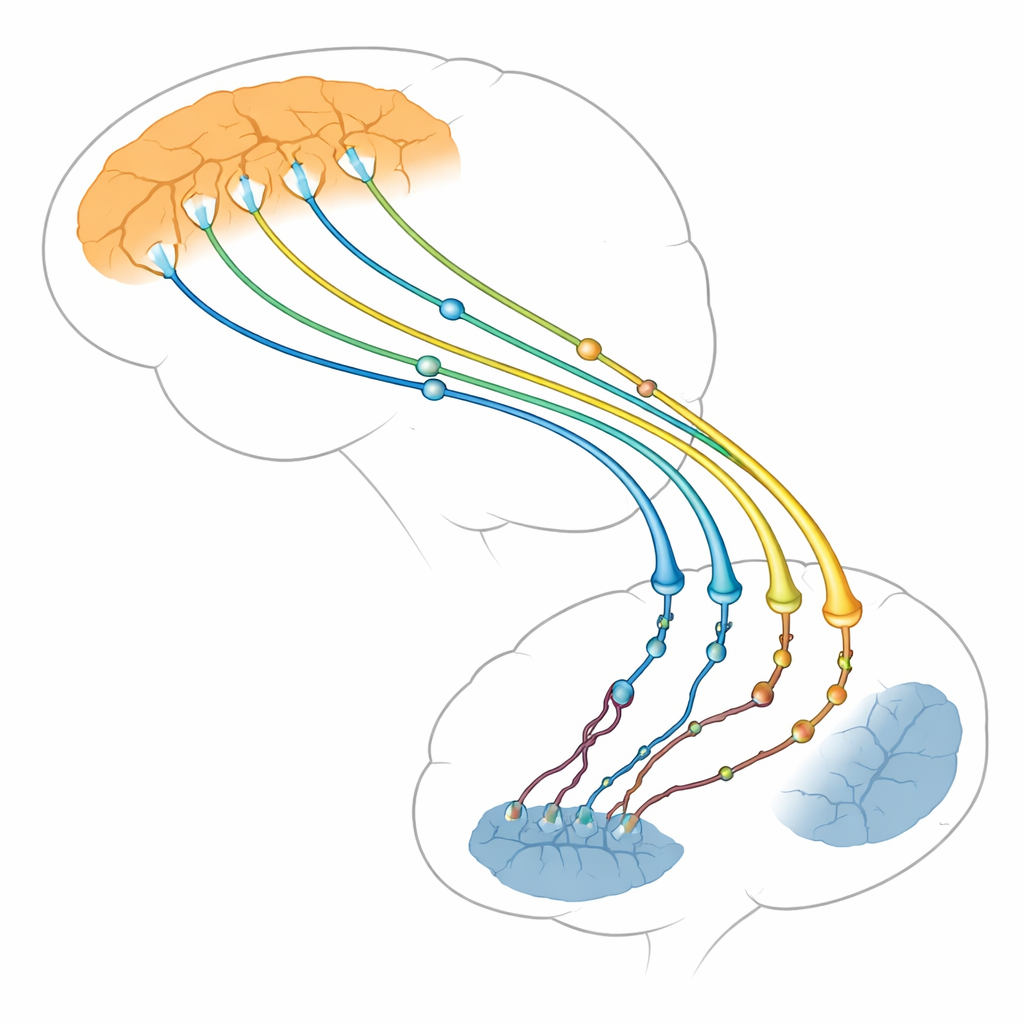
Huntingtin als Hauptorganisator an den Nervenenden
Huntingtin ist ein großes Protein, das in ganzen Nervenzellen vorkommt, aber besonders an Synapsen angereichert ist, wo es wie ein Gerüst oder Andockstation für mehr als 3.000 Partnerproteine wirkt. Auf der sendenden Seite der Synapse hilft es, Fracht – beispielsweise energieproduzierende Mitochondrien, mit Neurotransmittern gefüllte Vesikel und Wachstumsfaktor-Pakete wie BDNF – entlang innerer Schienen zur Nervenende zu transportieren. Es hilft auch, Vesikel so zu positionieren, dass sie freigesetzt werden können, sie mit der Membran zu verschmelzen, um ihren Inhalt abzugeben, und die Membran wieder aufzunehmen, um neue Vesikel zu recyceln. Der Artikel zeigt, wie Huntingtin über Adapter wie HAP1 und HIP1 und kleine Schaltproteine, die Rab-Proteine heißen, diesen ständigen Verkehr koordiniert, damit Nervenzellen schnell und zuverlässig Signale senden können.
Wenn mutiertes Huntingtin den Betrieb blockiert
Bei der Huntington-Krankheit verändert eine überlange Folge von Glutamin-Bausteinen in Huntingtin dessen Gestalt und Bindungsvorlieben subtil. Diese mutierte Form haftet an manchen Partnern zu stark und an anderen zu schwach. Infolgedessen fragmentieren und verharren Mitochondrien im Zellkörper statt die Nervenenden zu erreichen, BDNF-Pakete bewegen sich langsamer und in die falsche Richtung, und Vesikel sind nicht richtig bestückt, verankert oder recycelt. Wichtige Helferproteine können in Huntingtin-reichen Klumpen gefangen werden, und das Reinigungssystem, das beschädigte Komponenten entfernen sollte – eine spezialisierte Form des zellulären Selbstabbaus, Autophagie genannt – arbeitet weniger effizient. Mit der Zeit wird die sendende Seite der kortiko-striatalen Synapse energiell, wachstumsfördernd und vesikelmäßig ausgehungert, was zu nachlassenden Signalen und schließlich zur Trennung führt.
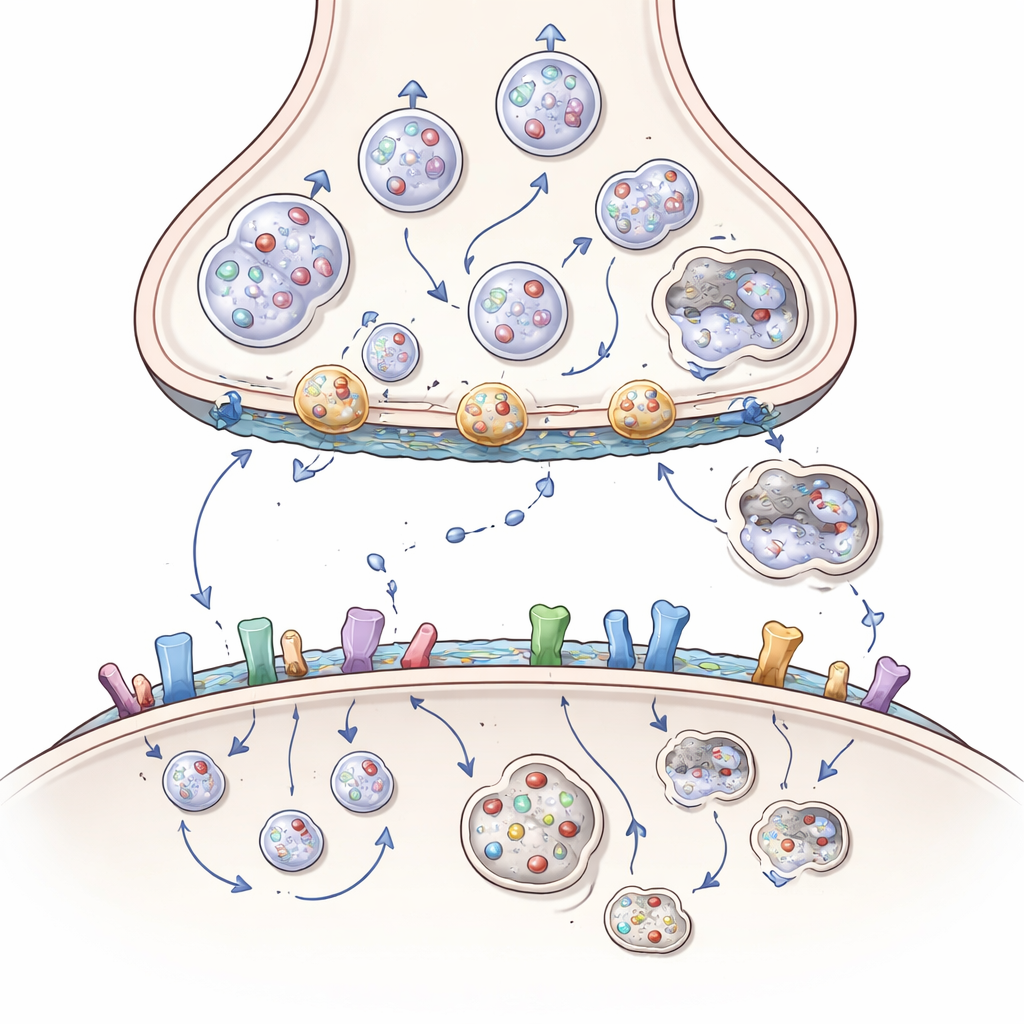
Signalempfang, Klebrigkeit und die Rolle von Lipiden
Der Schaden beschränkt sich nicht auf die sendende Seite. Auf der empfangenden Seite hilft Huntingtin zu steuern, wie viele Rezeptoren für erregende und hemmende Botenstoffe an der Oberfläche sitzen, wo sie sich zusammenlagern und wie schnell sie recycelt werden. Indem es mit Gerüstproteinen und Enzymen zusammenarbeitet, die Fettschwänze anhängen, hält Huntingtin normalerweise bestimmte Glutamatrezeptoren an schützenden synaptischen Positionen und begrenzt schädlichere, extrasynaptische Varianten. Mutiertes Huntingtin stört dieses Gleichgewicht und verschiebt Rezeptoren an Orte und in Kombinationen, die zu toxischem Calciumeinstrom und Zelltod neigen. Es beeinträchtigt außerdem Zelladhäsionsmoleküle, die Synapsen zusammenhalten, sowie Hirnlipide wie Cholesterin und Ganglioside, die Membranen formen und Signalübertragung unterstützen. Der Verlust dieser Lipide bei der Huntington-Krankheit schwächt Synapsen und Wachstumsfaktor-Signale zusätzlich.
Neue Ansatzpunkte für Therapien
Die Übersicht schließt mit der Feststellung, dass Huntingtin nicht nur ein passives Opfer der Mutation ist, sondern ein aktiver Organisator von Synapsen, dessen Partnerschaften in der Krankheit entgleisen. Da viele seiner wichtigsten Verbündeten auf der präsynaptischen Seite besonders anfälliger kortikaler Neurone sitzen, könnte das gezielte Ansprechen dieser Partner – etwa Transportadapter, Wachstumsfaktor-Transporteure, Schlüsselenzyme wie ADAM10 oder Lipidwege – neue Wege eröffnen, um kortiko-striatale Synapsen zu schützen, ohne Huntingtin vollständig entfernen zu müssen. Fortschritte in der Super-Auflösungsbildgebung, in „Brain-on-a-chip“-Modellen und in der Untersuchung chemischer Modifikationen von Huntingtin könnten künftig genau zeigen, wo und wann ein Eingreifen sinnvoll ist, und damit Hoffnung auf Therapien wecken, die die Kommunikationswege des Gehirns bewahren und das Fortschreiten der Huntington-Krankheit verlangsamen.
Zitation: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Schlüsselwörter: Huntington-Krankheit, kortiko-striatale Synapse, Huntingtin-Protein, synaptische Dysfunktion, axonaler Transport