Clear Sky Science · fr
Huntingtine et ses alliés au synapse cortico-striatal
Pourquoi cette protéine cérébrale compte
La maladie de Huntington est surtout connue pour provoquer des mouvements saccadés et des troubles de la mémoire, mais en son cœur il s’agit d’une maladie de la communication rompue entre les cellules nerveuses. Cet article examine comment une seule protéine, la huntingtine, et ses nombreux « alliés » aux jonctions entre neurones contribuent à maintenir le fonctionnement des circuits cérébraux — et comment les altérations de cette protéine font dérailler ces circuits, en particulier le long d’une voie clé reliant la pensée au mouvement. Comprendre ce câblage caché offre de nouvelles pistes pour ralentir ou prévenir la maladie.
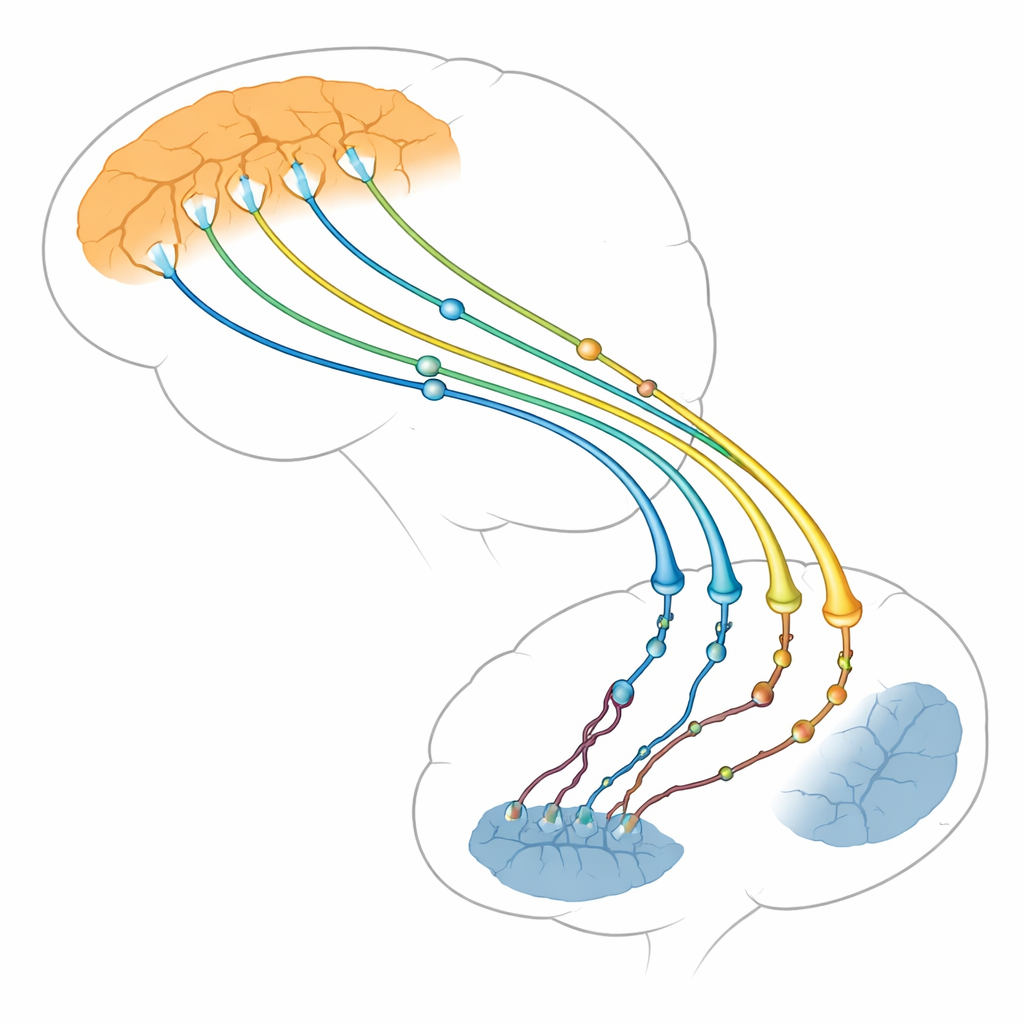
Le carrefour cérébral de la pensée et du mouvement
Les auteurs se concentrent sur la connexion entre deux régions du cerveau : la couche externe pensante (cortex) et une structure profonde qui aide à contrôler les actions et les habitudes (striatuм). Les neurones du cortex envoient de longues fibres vers le striatum, où elles forment des milliers de points de contact minuscules, ou synapses. Ces synapses cortico-striatales sont parmi les premières structures à faiblir dans la maladie de Huntington, bien avant la mort de nombreux neurones. Les images cérébrales et les études animales montrent que lorsque cette voie s’affaiblit, les symptômes s’aggravent. La revue soutient que les problèmes ne surviennent pas seulement du côté récepteur striatal, comme on le pensait autrefois, mais aussi du côté cortical émetteur, qui peut en réalité être à l’origine d’une grande partie des dégâts.
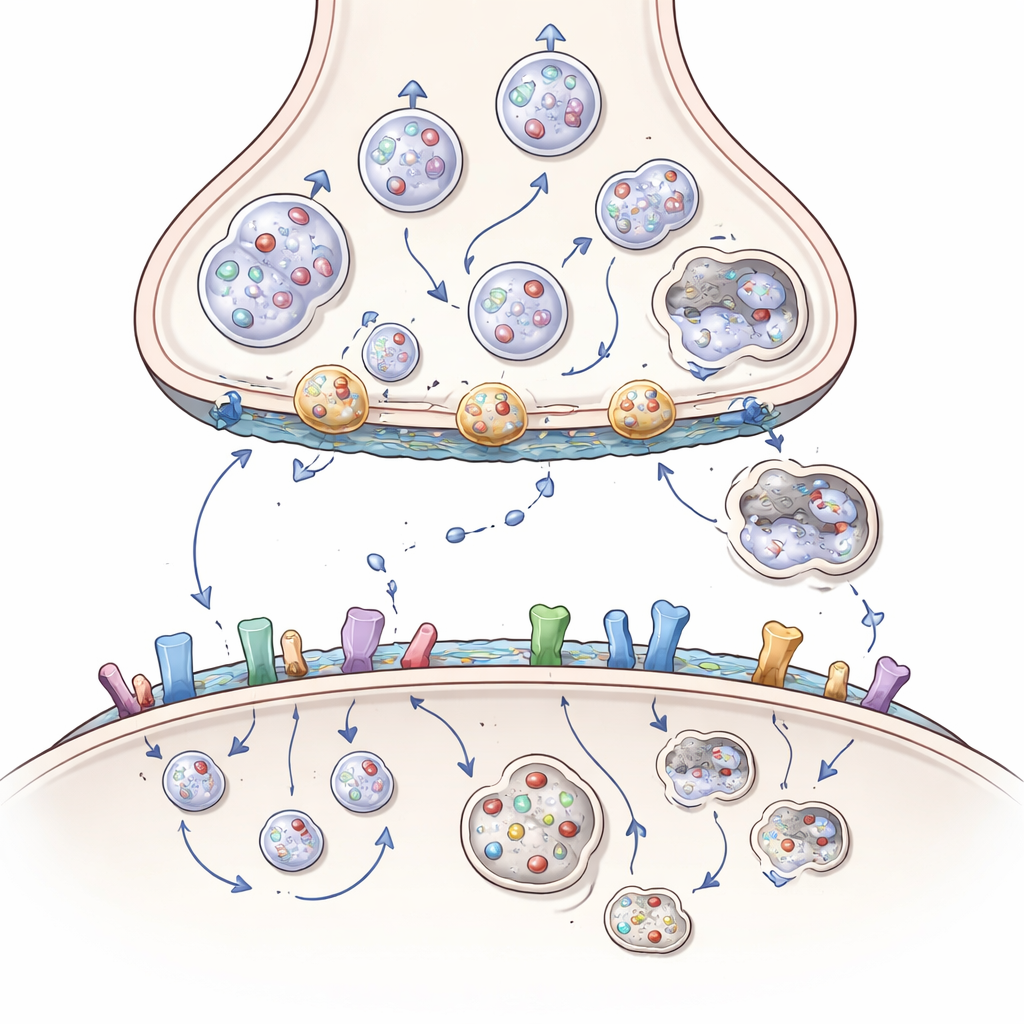
La huntingtine, organisatrice des terminaisons nerveuses
La huntingtine est une grande protéine présente dans l’ensemble des neurones, mais elle est particulièrement abondante aux synapses, où elle agit comme un échafaudage ou une station d’accostage pour plus de 3 000 protéines partenaires. Du côté pré-synaptique, elle aide à déplacer des cargaisons — telles que des mitochondries productrices d’énergie, des vésicules remplies de neurotransmetteurs et des paquets de facteurs de croissance appelés BDNF — le long de voies internes jusqu’à l’extrémité nerveuse. Elle aide aussi à positionner les vésicules pour leur libération, à les fusionner avec la membrane pour déverser leur contenu, puis à récupérer la membrane pour recycler de nouvelles vésicules. L’article montre comment la huntingtine, via des adaptateurs comme HAP1 et HIP1 et de petits interrupteurs appelés protéines Rab, coordonne ce trafic constant pour que les neurones puissent transmettre des signaux rapidement et de manière fiable.
Quand la huntingtine mutante bloque la machinerie
Dans la maladie de Huntington, une longue séquence supplémentaire de glutamines dans la huntingtine modifie subtilement sa conformation et ses affinités de liaison. Cette forme mutante s’attache trop fortement à certains partenaires et trop faiblement à d’autres. En conséquence, les mitochondries se fragmentent et restent bloquées dans le corps cellulaire au lieu d’atteindre les terminaisons nerveuses, les paquets de BDNF voyagent plus lentement et dans la mauvaise direction, et les vésicules ne sont pas correctement approvisionnées, ancrées ou recyclées. Des protéines auxiliaires clés peuvent se retrouver piégées dans des amas riches en huntingtine, et le système de « nettoyage » chargé d’éliminer les composants endommagés — une version spécialisée de l’auto-digestion cellulaire appelée autophagie — fonctionne moins efficacement. Avec le temps, le côté pré-synaptique de la synapse cortico-striatale s’appauvrit en énergie, en soutien par les facteurs de croissance et en vésicules fraîches, conduisant à l’affaiblissement des signaux et à une déconnexion éventuelle.
Réception du signal, adhérence et rôle des lipides
Les dommages ne se limitent pas au côté émetteur. Du côté récepteur, la huntingtine aide à contrôler combien de récepteurs pour les messagers excitateurs et inhibiteurs se trouvent à la surface, où ils se regroupent et à quelle vitesse ils sont recyclés. En s’associant à des protéines d’échafaudage et à des enzymes qui ajoutent des queues grasses, la huntingtine maintient normalement certains récepteurs au glutamate dans des positions synaptiques protectrices et limite les récepteurs extrasynaptiques plus nocifs. La huntingtine mutante perturbe cet équilibre, poussant les récepteurs vers des localisations et des combinaisons qui favorisent un afflux toxique de calcium et la mort cellulaire. Elle interfère également avec les molécules d’adhésion cellulaire qui maintiennent les synapses ensemble et avec des lipides cérébraux tels que le cholestérol et les gangliosides qui façonnent les membranes et soutiennent la signalisation. La perte de ces lipides dans la maladie de Huntington affaiblit encore les synapses et la signalisation par facteurs de croissance.
Nouveaux axes pour la thérapie
La revue conclut que la huntingtine n’est pas seulement une victime passive de la mutation mais une organisatrice active des synapses dont les partenariats déraillent dans la maladie. Parce que beaucoup de ses alliés les plus critiques se trouvent du côté présynaptique des neurones corticaux vulnérables, cibler ces partenaires — tels que les adaptateurs de transport, les transporteurs de facteurs de croissance, des enzymes clés comme ADAM10 ou les voies lipidiques — pourrait offrir de nouvelles voies pour protéger les synapses cortico-striatales sans avoir à éliminer complètement la huntingtine. Les progrès futurs en imagerie super-résolution, en modèles « brain-on-a-chip » et dans l’étude des modifications chimiques de la huntingtine pourraient révéler précisément où et quand intervenir, nourrissant l’espoir de thérapies qui préservent les lignes de communication du cerveau et ralentissent la progression de la maladie de Huntington.
Citation: Zuccato, C., Scolz, A. & Iennaco, R. Huntingtin and its allies at the cortico-striatal synapse. Cell Death Dis 17, 412 (2026). https://doi.org/10.1038/s41419-026-08584-6
Mots-clés: Maladie de Huntington, synapse cortico-striatale, protéine huntingtine, dysfonction synaptique, transport axonal