Clear Sky Science · pl
Integracja, wyrównanie i adnotacja danych pojedynczych komórek RNA-seq z wielu gatunków przy użyciu CAMEX
Dlaczego te badania są ważne
Każde ciało zwierzęce zbudowane jest z bogatej gamy typów komórek, a mimo to wciąż brakuje nam przejrzystej mapy tego, jak te komórki porównują się między gatunkami i jak zmieniały się w toku ewolucji. W tym badaniu wprowadzono CAMEX, narzędzie obliczeniowe, które skleja dane o aktywności genów w pojedynczych komórkach z wielu różnych zwierząt w wspólny obraz. Dla czytelnika nietechnicznego jest to ekscytujące, ponieważ przybliża nas do odpowiedzi na pytania, które typy komórek są naprawdę uniwersalne, które są unikatowe dla ludzi, oraz jak organy takie jak mózg, wątroba i jądra kształtowały się w czasie ewolucji.
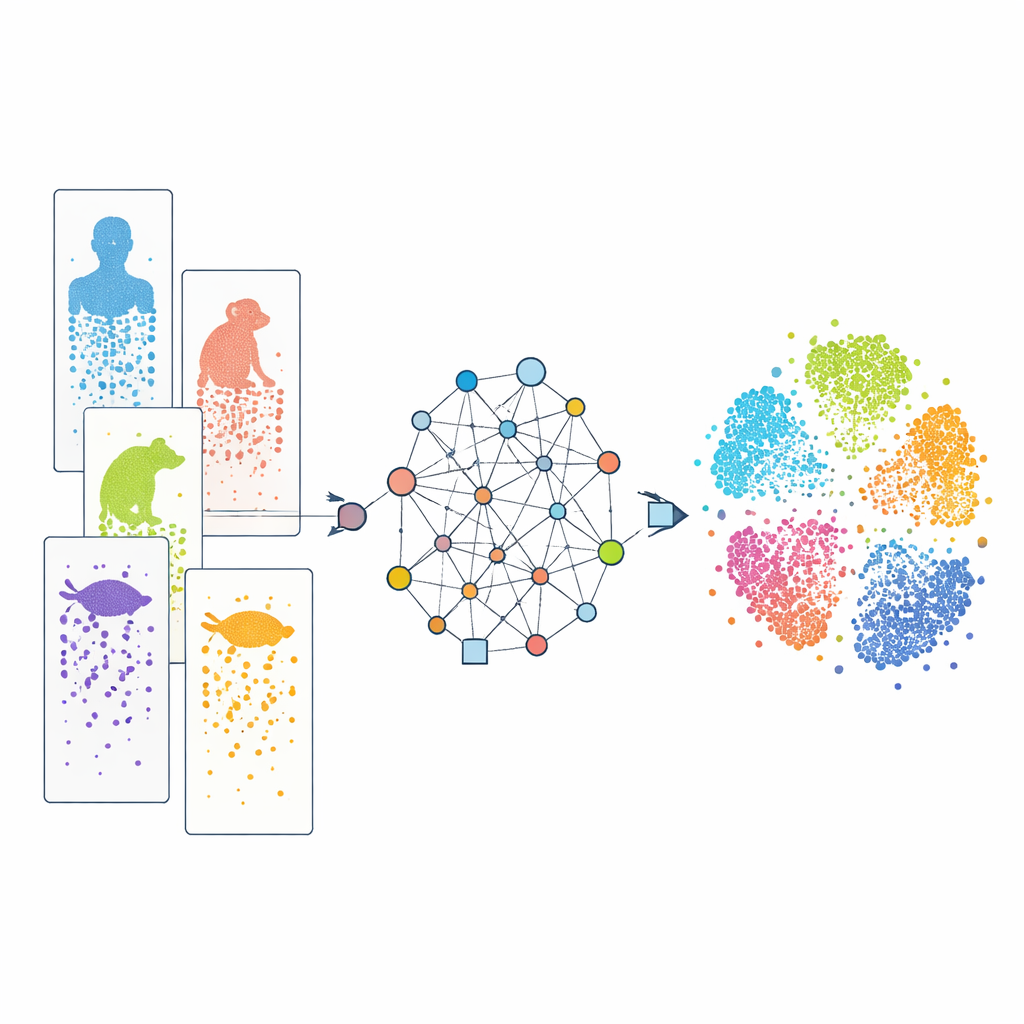
Patrząc na komórki pojedynczo
Nowoczesne sekwencjonowanie RNA pojedynczych komórek pozwala naukowcom odczytać, które geny są aktywne w tysiącach do milionów poszczególnych komórek w jednym eksperymencie. Porównując te wzorce, badacze mogą sortować komórki na typy i śledzić ich rozwój. Istnieje już wiele takich zbiorów danych dla ludzi, naczelnych, myszy, ryb, gadów i innych. Jednak każde badanie często używa różnych technologii eksperymentalnych, a gatunki różnią się zestawami genów. Do tego nasza wiedza o genach jest nierówna: dobrze zbadane zwierzęta laboratoryjne mają znacznie lepsze adnotacje niż rzadkie gatunki. Te różnice działają jak „efekty partii” i niekompletne słowniki, co utrudnia dopasowanie podobnych komórek między gatunkami i rozróżnienie, które cechy są naprawdę wspólne, a które specyficzne dla danego gatunku.
Grafowe podejście do łączenia gatunków
CAMEX pokonuje te przeszkody, przekształcając wszystkie dane w jedną dużą sieć, która obejmuje zarówno komórki, jak i geny. W tej sieci komórki łączą się z genami, które wyrażają, z najbardziej podobnymi sąsiednimi komórkami, a geny łączone są między gatunkami, jeśli oceniono, że są ze sobą powiązane ewolucyjnie — nawet gdy relacja jest wiele-do-wielu, a nie proste jeden-do-jednego. Specjalistyczny typ modelu uczenia maszynowego, heterogeniczna grafowa sieć neuronowa, przekazuje informacje wzdłuż tych połączeń i uczy się zwartej „osadzającej” reprezentacji każdej komórki i genu we wspólnej przestrzeni o niskim wymiarze. Dla integracji danych model jest trenowany tak, by rekonstruować zarówno strukturę sieci, jak i oryginalne wzorce aktywności genów, bez uprzedniego informowania o typach komórek. Dla adnotacji komórek ten sam kodownik zasila klasyfikator oparty na mechanizmie attention, który potrafi przenosić znane etykiety z gatunku referencyjnego na mniej zbadane.
Odkrywanie wspólnych typów komórek i rozwoju
Autorzy pokazują, że CAMEX przewyższa zestaw popularnych narzędzi, gdy jest testowany na wymagających, rzeczywistych zestawach danych. W danych z wątroby, jajnika i trzustki obejmujących do czterech gatunków i wiele platform eksperymentalnych, CAMEX najlepiej zrównoważył dwa konkurencyjne cele: usuwanie sztucznych różnic między partiami przy zachowaniu prawdziwych biologicznych rozróżnień między typami komórek. Dokładnie wyrównał powszechne populacje komórek, takie jak hepatocyty i komórki odpornościowe, a co ważne — zachował rzadkie typy komórek, które inne metody miały tendencję zlewać. W dramatycznym teście CAMEX zintegrował dane z jąder od 11 gatunków, od naczelnych po dziobaka i kurę. Odtworzył ciągłą ścieżkę, w której komórki rozrodcze dojrzewają do plemników, i wykazał, że użycie relacji genów wiele-do-wielu jest kluczowe dla utrzymania wydajności wraz ze wzrostem ewolucyjnej odległości między gatunkami. Model skutecznie wyrównał też etapy rozwoju organów w siedmiu gatunkach, rozszerzając koncepcję klasycznych stadiów rozwojowych Carnegie poza niewielki zestaw organizmów modelowych, dla których pierwotnie je zdefiniowano.
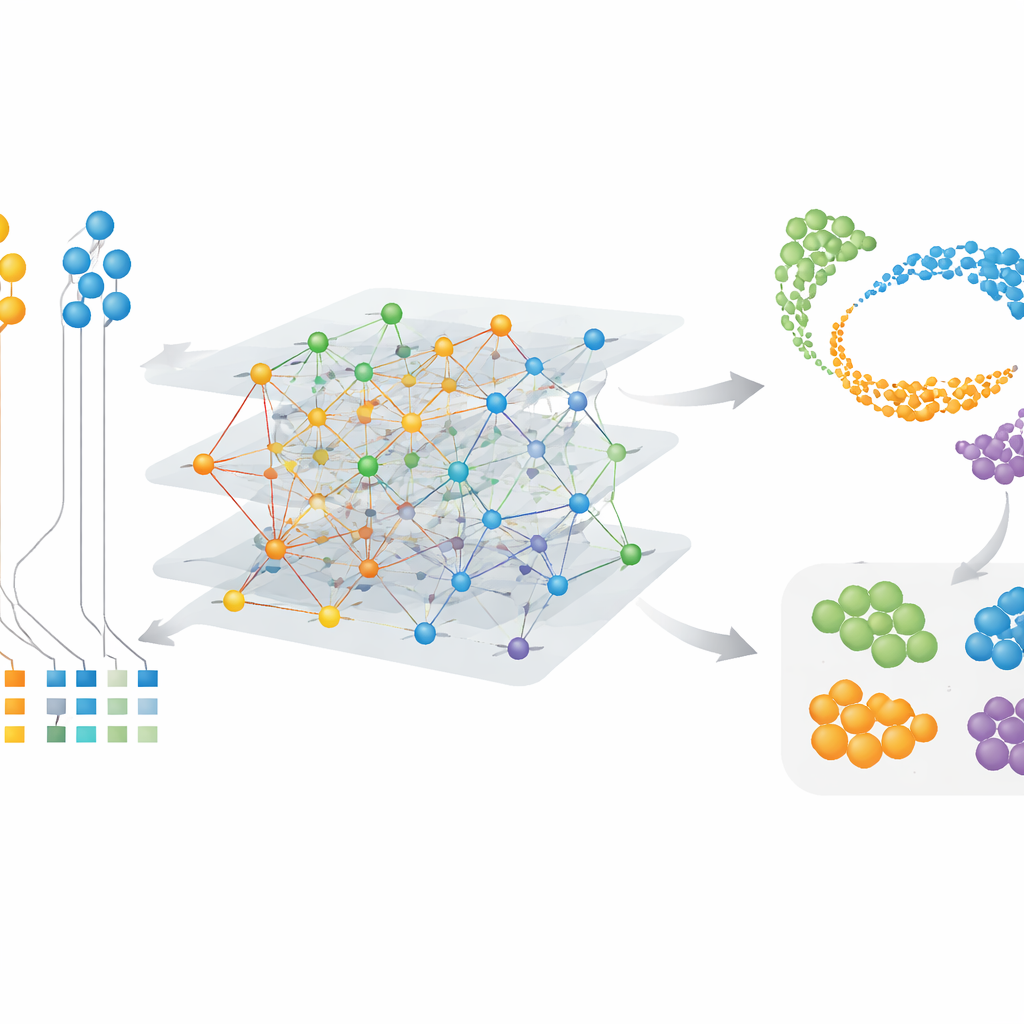
Wykrywanie komórek i modułów genowych specyficznych dla gatunków
Ponieważ CAMEX uczy osadzeń zarówno dla komórek, jak i genów, potrafi podkreślić cechy szczególne, a nie tylko wspólne. W zbiorach danych mózgowych obejmujących człowieka, mysz, jaszczurkę i żółwia, CAMEX zintegrował dane i przy wykorzystaniu ludzkich etykiet jako wskazówki dokładnie adnotował typy komórek w innych gatunkach, nawet małe podgrupy, takie jak perycyty mózgowe u żółwia. Zastosowany do szczegółowej mapy grzbietowo-bocznej kory przedczołowej naczelnych, autorzy byli w stanie wyodrębnić konkretne podtypy mikrogleju — komórek odpornościowych mózgu — które występują tylko u ludzi lub dzielone są z szympansami. Grupując osadzenia genów, znaleźli też zbiory genów powiązane z kluczowymi funkcjami: na przykład moduły aktywne w somatycznych komórkach podporowych w jądrach oraz inne związane z mejozą, procesem podziału komórkowego prowadzącym do powstawania plemników. Wyniki te wskazują zarówno na zachowane programy, jak i gatunkowo specyficzne modyfikacje zachowania komórek.
Co to znaczy w szerszym kontekście
Mówiąc prosto, CAMEX to potężny nowy „silnik translacyjny” dla danych pojedynczych komórek w całym drzewie życia. Pomaga naukowcom zobaczyć, kiedy komórki z różnych zwierząt wykonują w zasadzie tę samą funkcję, kiedy się rozeszły i jak porównują się linie czasowe rozwoju między gatunkami. Choć metoda ma nadal ograniczenia — takie jak poleganie na istniejących mapach homologii i ogólne trudności w interpretacji modeli grafowych — to już umożliwia bogatsze porównania ewolucyjne niż było to wcześniej możliwe. Z czasem narzędzia takie jak CAMEX mogą pomóc w zbudowaniu prawdziwego filogenetycznego drzewa typów komórek, dopracować modele rozwoju organów i ukierunkować poszukiwania typów komórek istotnych w chorobach oraz celów lekowych zarówno u ludzi, jak i w modelach zwierzęcych.
Cytowanie: Guo, ZH., Huang, DS. & Zhang, S. Multi-species integration, alignment and annotation of single-cell RNA-seq data with CAMEX. Nat Commun 17, 3017 (2026). https://doi.org/10.1038/s41467-026-69696-3
Słowa kluczowe: sekwencjonowanie RNA pojedynczych komórek, integracja międzygatunkowa, grafowe sieci neuronowe, ewolucja typów komórek, genomika porównawcza