Clear Sky Science · it
Una variante biallelica di MRPL42 causa una sindrome da deficit combinato della fosforilazione ossidativa rivelata dal multi‑omico
Quando le centrali energetiche della cellula vacillano
I mitocondri sono spesso chiamati le centrali elettriche delle nostre cellule, trasformando silenziosamente il combustibile nell’energia che mantiene il cuore che batte e il cervello che pensa. Questo articolo racconta la storia di una neonata la cui grave malattia ha portato gli scienziati a scoprire un piccolo difetto nella macchina di sintesi proteica all’interno dei mitocondri. Combinando diverse tecniche di laboratorio all’avanguardia, i ricercatori mostrano come una singola alterazione ereditaria in un gene possa destabilizzare la produzione di energia cellulare e causare una malattia multisistemica fatale.
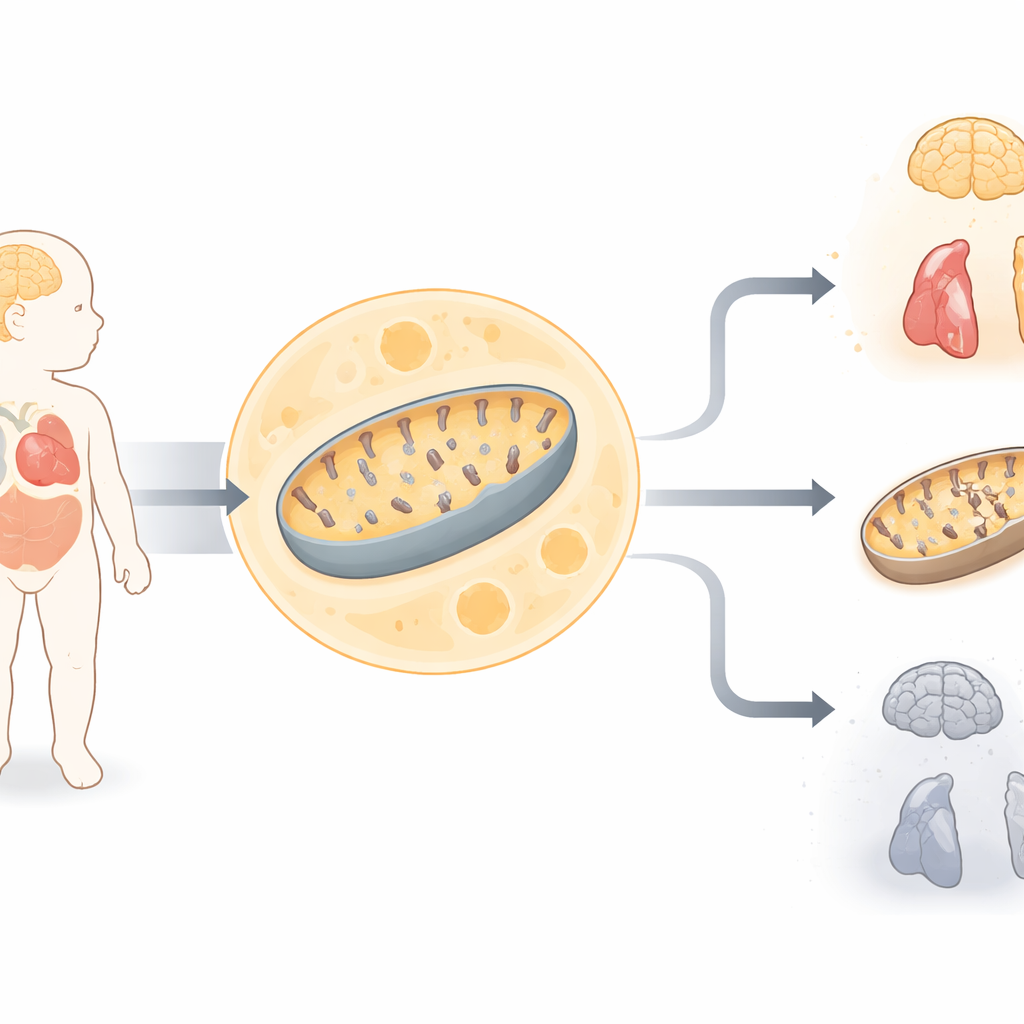
Una neonato in serie difficoltà
Lo studio si concentra su una bambina nata da genitori consanguinei che, a prima vista, sembrava normale ma sviluppò rapidamente gravi problemi medici. Presentava debolezza muscolare, difficoltà respiratorie, crisi epilettiche, problemi uditivi, alterazioni cardiache e livelli elevati di acido lattico nel sangue — un segnale che le cellule stanno faticando a produrre energia. Le scansioni cerebrali rivelarono una perdita diffusa di tessuto cerebrale e i test su cellule della pelle mostrarono che due grandi complessi enzimatici produttori di energia nei suoi mitocondri funzionavano al di sotto del normale. Questi indizi indicavano un grave disturbo del metabolismo energetico mitocondriale, ma i test genetici standard eseguiti prima della nascita non avevano rilevato una causa nota.
Alla ricerca di un errore nascosto nel gene
Per andare oltre i test di routine, il team adottò una strategia “multi‑omica”: leggere il DNA, i messaggi di RNA copiati dal DNA e l’insieme completo delle proteine nelle cellule della bambina. Il sequenziamento dell’intero genoma rivelò una variante rara in entrambe le copie di un gene chiamato MRPL42, che codifica un piccolo componente della grande unità di sintesi proteica (ribosoma) all’interno dei mitocondri. Da sola, questa modifica del DNA era classificata come incerta perché MRPL42 non era stato precedentemente associato a malattie umane. Tuttavia, il sequenziamento dell’RNA mostrò che il messaggio genico alterato era per lo più missplicato: una sezione critica veniva saltata in circa quattro molecole su cinque, causando uno scorrimento del frame di lettura e un segnale di stop precoce. Solo una piccola frazione di messaggio normale, e quindi una parte di proteina normale, veniva ancora prodotta.
Come un pezzo mancante indebolisce la centrale
Studi proteomici e di imaging sulle cellule della pelle della paziente hanno rivelato cosa significasse in pratica questo messaggio difettoso. I livelli della proteina MRPL42 risultarono ridotti a circa un quarto del normale e la rete mitocondriale appariva più frammentata rispetto alle cellule sane. Indagini proteiche dettagliate mostrarono che molti componenti costitutivi del ribosoma mitocondriale — sia della sua subunità grande sia di quella piccola — erano diminuiti, suggerendo che l’intera struttura era meno stabile. Parallelamente, numerosi componenti dei complessi produttori di energia I e IV, e in misura minore del complesso III, risultarono ridotti, mentre il complesso II rimase invariato. Misurazioni dell’utilizzo di ossigeno confermarono che la capacità delle cellule di respirare e produrre ATP, la moneta energetica cellulare, era fortemente compromessa.
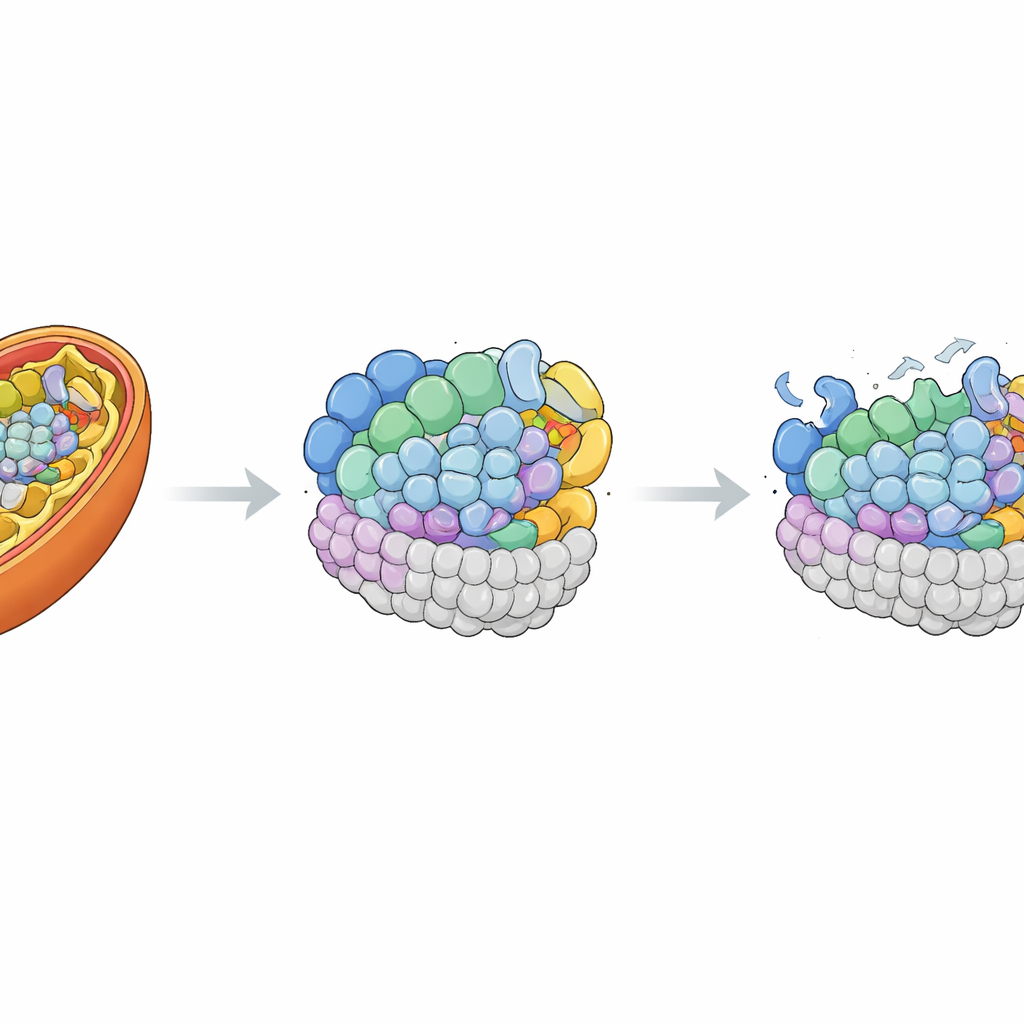
Ripristinare il pezzo mancante
Per dimostrare che il difetto in MRPL42 era effettivamente responsabile, i ricercatori reintrodussero una copia normale del gene nelle cellule della paziente utilizzando un vettore virale innocuo. Questo aumentò i livelli di MRPL42 oltre quelli delle cellule di controllo e ripristinò parzialmente la normale forma tubulare della rete mitocondriale. Importante, molti dei componenti proteici persi del ribosoma mitocondriale e dei complessi I e IV ritornarono verso quantità normali. Quando il team misurò di nuovo la respirazione mitocondriale, trovò che la capacità massima di respirazione e la capacità di riserva quasi si normalizzarono, e anche il turnover energetico di base migliorò. Questi esperimenti di salvataggio collegano fortemente i difetti cellulari della bambina alla carenza di MRPL42 funzionale.
Cosa significa per i pazienti e per la scienza
Nel complesso, lo studio dimostra che una variante loss‑of‑function biallelica (a due copie) in MRPL42 può causare un disturbo grave e spesso fatale in cui più complessi energetici mitocondriali falliscono. Il gene sembra non essere assolutamente essenziale — si forma ancora una certa proteina, permettendo lo sviluppo fino alla nascita — ma è cruciale per mantenere il ribosoma mitocondriale sufficientemente stabile da sostenere una produzione energetica normale. Per le famiglie, questo lavoro fornisce una spiegazione molecolare per una malattia devastante e apre la strada a una consulenza genetica accurata. Più in generale, sottolinea quanto l’integrazione profonda di sequenziamento del DNA, analisi dell’RNA e profilazione proteica possa svelare nuovi geni di malattia e chiarire come piccole anomalie nella macchina cellulare si propaghino fino a compromettere interi organi e, in ultima istanza, l’intero organismo.
Citazione: Boschann, F., Kopp, J., Römer, S. et al. A biallelic MRPL42 variant causes a combined oxidative phosphorylation deficiency syndrome revealed by multi-omics. npj Genom. Med. 11, 20 (2026). https://doi.org/10.1038/s41525-026-00564-1
Parole chiave: malattia mitocondriale, fosforilazione ossidativa, proteina ribosomiale, multi‑omico, variante genetica