Clear Sky Science · it
Diagnosi genetica della CAH legata a CYP21A2: il sequenziamento long-read con adaptive sampling è una soluzione accurata e scalabile
Perché è importante per famiglie e medici
Molti neonati vengono oggi sottoposti allo screening alla nascita per l’iperplasia surrenalica congenita, un disturbo ormonale che può essere potenzialmente mortale se non riconosciuto. Tuttavia confermare la diagnosi a livello di DNA è sorprendentemente difficile, perché il gene chiave si trova in una porzione del genoma confusa, affiancato da un «gemello» quasi identico che funge da inganno. Questo studio dimostra che una nuova modalità di lettura del DNA, chiamata sequenziamento long-read con adaptive sampling, può fare chiarezza, offrendo risposte più rapide e affidabili per i pazienti e le loro famiglie.
Un disturbo ormonale con complessità genetiche
L’iperplasia surrenalica congenita (CAH) influisce su come le ghiandole surrenali producono ormoni essenziali che regolano l’equilibrio salino, le risposte allo stress e i livelli di ormoni sessuali. In circa il 95% dei casi, il problema è riconducibile a variazioni in un singolo gene, CYP21A2, che codifica per l’enzima 21‑idrossilasi. Se quell’enzima è assente o poco efficiente, l’organismo non riesce a produrre abbastanza cortisolo e aldosterone e, al contrario, sovraproduce androgeni, con manifestazioni che vanno da crisi da perdita di sale nei neonati a ipertricosi e problemi di fertilità in età adulta. La maggior parte dei pazienti porta varianti patogene diverse su ciascuna copia del gene, e la combinazione di queste varianti influenza fortemente la gravità della malattia.
Un gene nascosto accanto al suo sosia
La difficoltà principale è che CYP21A2 si trova in un quartiere complesso sul cromosoma 6, accanto a un «pseudogene» quasi identico, CYP21A1P, che non è più funzionale. Il loro codice DNA è circa il 98% sovrapponibile, e la regione è soggetta a eventi di rimescolamento in cui pezzi del gene funzionante e dello pseudogene si scambiano, si fondono o vengono eliminati. I metodi tradizionali si basano sull’amplificazione di tratti specifici di DNA mediante PCR seguita dalla lettura con sequenziamento Sanger, talvolta integrata con una tecnica chiamata MLPA per contare le copie geniche. Queste metodiche sono lente, laboriose e possono avere difficoltà a rilevare riorganizzazioni di grandi dimensioni o a stabilire se più varianti sono sullo stesso cromosoma o su cromosomi opposti—informazioni cruciali per valutare il rischio nei pazienti e nei familiari.
Un nuovo modo di leggere la regione problematica
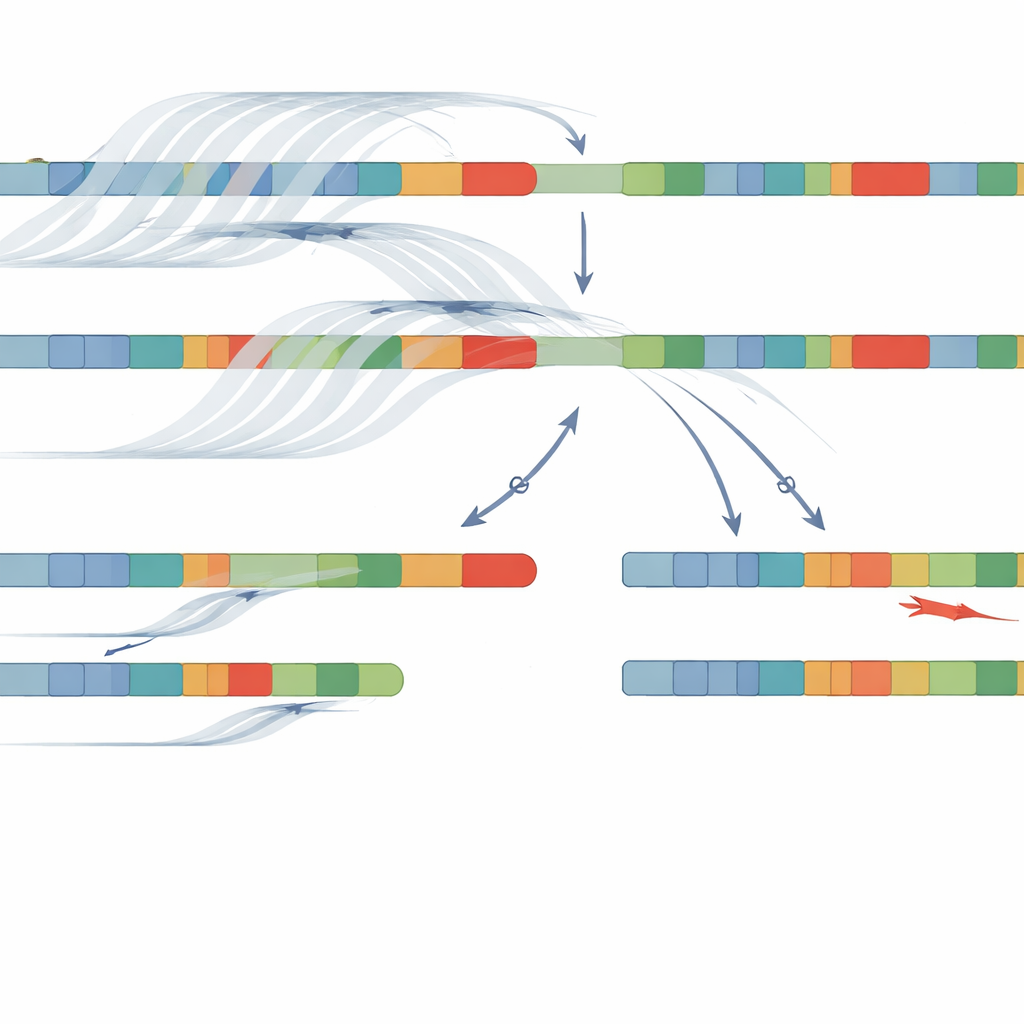
I ricercatori hanno esplorato una strategia alternativa usando il sequenziamento long-read sulla piattaforma Oxford Nanopore. Invece di frammentare il DNA in pezzi molto corti, questa tecnologia legge tratti molto più lunghi, agevolando la connessione di elementi distanti lungo il cromosoma. Hanno impiegato l’«adaptive sampling», in cui il sequenziatore viene istruito in tempo reale a concentrarsi su una regione scelta—in questo caso il cromosoma 6—aumentando la copertura senza passaggi di laboratorio complessi. Da 34 pazienti con diagnosi clinica di CAH, il DNA è stato sequenziato senza PCR, evitando bias che possono far scomparire una versione del gene dalla rilevazione. Per gestire l’estrema somiglianza tra CYP21A2 e il suo pseudogene, il gruppo ha sviluppato un software personalizzato chiamato NanoCAH, che analizza con cura come ogni lettura long-read si allinea, decide se proviene dal gene funzionante o dall’inganno e ricostruisce le due copie della regione per ciascun paziente.
Risposte più chiare nella maggior parte dei pazienti
Combinando l’adaptive sampling, il sequenziamento long-read e NanoCAH, il team ha identificato le varianti causali in 32 dei 34 pazienti, con un tasso di successo del 94%. Non solo hanno confermato quasi tutti i riscontri ottenuti con metodi precedenti, ma hanno anche scoperto dettagli importanti che erano stati persi o classificati in modo errato. In alcuni pazienti, test precedenti avevano descritto una variante come «omozigote», come se entrambe le copie del cromosoma contenessero lo stesso errore; il nuovo approccio ha rivelato che una copia portava in realtà una delezione o una fusione complessa tra gene e pseudogene. In altri casi sono state identificate mutazioni puntiformi prima non osservate, grazie alla lettura dell’intera regione invece che di un insieme fisso di hotspot comuni. Fondamentale, le letture lunghe hanno permesso la «fase» delle varianti—mostrando quali cambiamenti sono presenti insieme sullo stesso cromosoma—senza necessità del DNA dei genitori.
Implicazioni per i test futuri
Lo studio conclude che il sequenziamento long-read con adaptive sampling, abbinato a software di analisi specializzati, può fornire diagnosi genetiche per la CAH più rapide, scalabili e accurate rispetto ai test standard attuali. Fornendo un quadro più chiaro delle complesse riorganizzazioni geniche e separando in modo affidabile il gene reale dal suo quasi‑gemello, questo approccio può migliorare la diagnosi, guidare le scelte terapeutiche e supportare test di portatori e diagnostica prenatale più precisi nelle famiglie interessate. Poiché molti altri geni associati a malattie si trovano in tratti del DNA ripetuti e complessi, la stessa strategia potrebbe aiutare a risolvere questioni genetiche difficili ben oltre la CAH.
Citazione: Lildballe, D.L., Huno, M.R., Ridder, L.O.R. et al. Genetic diagnosis of CYP21A2-related CAH: adaptive sampling long-read sequencing is an accurate and scalable solution. Eur J Hum Genet 34, 535–542 (2026). https://doi.org/10.1038/s41431-026-02019-8
Parole chiave: iperplasia surrenalica congenita, CYP21A2, sequenziamento long-read, diagnosi genetica, adaptive sampling