Clear Sky Science · en
METTL3-mediated fibroblast-like synoviocytes senescence promotes temporomandibular joint osteoarthritis progression
Why jaw joint wear and tear matters
Clicking, pain, or stiffness in the jaw can be more than an annoyance; for many people it signals temporomandibular joint osteoarthritis, a painful wear-and-tear disorder of the jaw joint. This study looks beneath the surface of the joint to ask a simple but important question: why does the cushioning cartilage in the jaw start to break down, and could slowing a particular kind of cell aging help protect it? The researchers uncover a molecular chain of events inside joint-lining cells that may open the door to more targeted, less invasive treatments than current drugs or surgery.
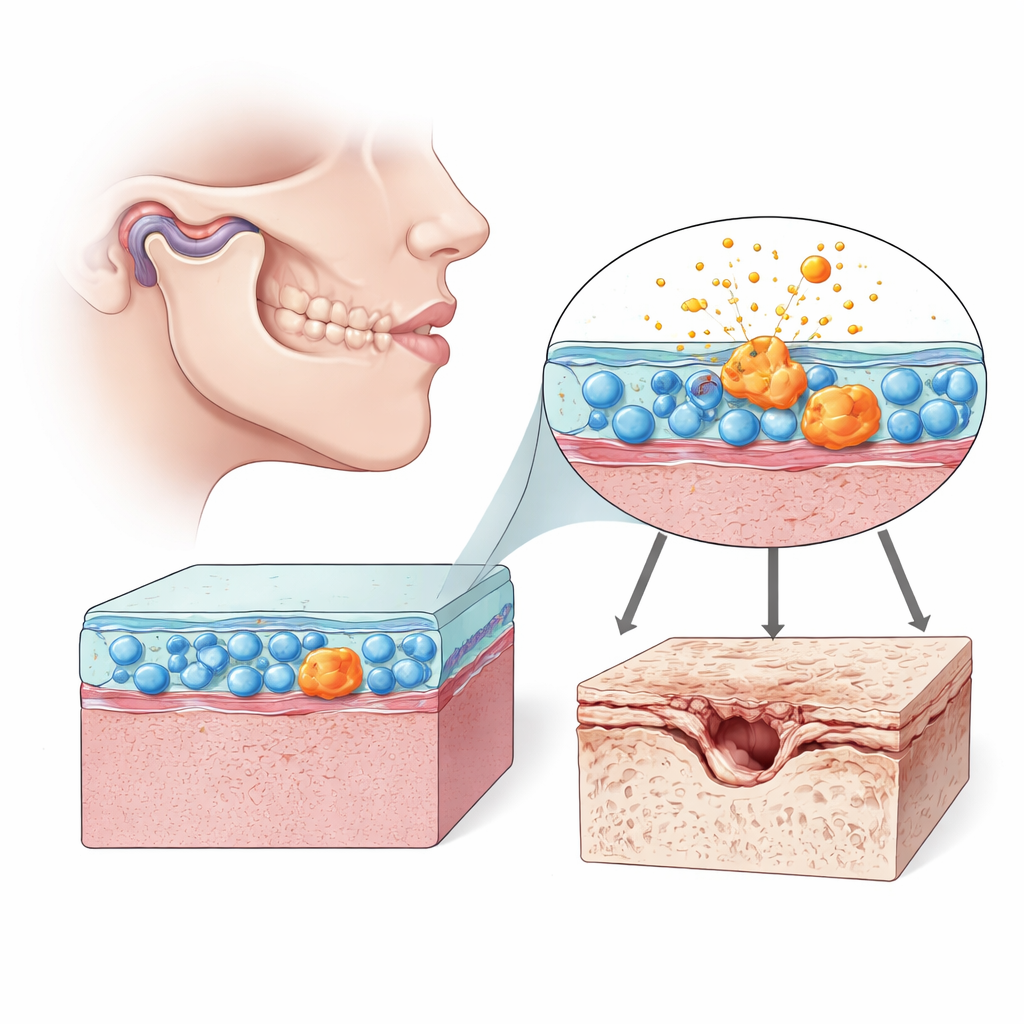
Cells that quietly shape joint health
Inside every movable joint is a soft lining called the synovium, packed with fibroblast-like synoviocytes—cells that help lubricate and nourish the joint. In the temporomandibular joint (TMJ), these cells help keep the tiny disc and cartilage surfaces gliding smoothly as we chew and talk. But when these synovial cells go awry, they can flood the joint with inflammatory molecules and enzymes that eat away at cartilage. The authors focused on how these cells behave in TMJ osteoarthritis in rats and in human cell cultures, paying special attention to signs of cellular aging, or senescence, and the health of their energy factories, the mitochondria.
When joint-lining cells grow old before their time
Using a chemical to trigger TMJ osteoarthritis in rats, the team confirmed classic features of the disease: roughened bone beneath the cartilage, thinning cartilage, more pain sensitivity, and higher levels of inflammatory messengers. In the joint lining, many synoviocytes showed markers of senescence—cells that have stopped dividing but remain metabolically active and highly inflammatory. These senescent cells had sluggish internal cleanup systems and damaged mitochondria. When the researchers isolated these joint-lining cells and grew them with human cartilage cells, the cartilage cells began producing more breakdown enzymes and less structural collagen, showing that the aged synovial cells could directly drive cartilage damage.
Broken cellular cleanup and a key molecular switch
Healthy cells constantly recycle worn-out mitochondria through a process called mitophagy, preventing buildup of defective components. In both rat and human synoviocytes pushed into a senescent state, mitophagy was weakened: damaged mitochondria piled up, mitochondrial membrane potential fell, and proteins linked to mitochondrial recycling declined. A drug that boosts mitophagy partially reversed these aging features, suggesting that poor mitochondrial cleanup helps lock these cells into a harmful, senescent state. The team then homed in on a protein called METTL3, which adds a chemical tag (m6A) to RNA molecules and can change how long those messages last in the cell. METTL3 levels were elevated in diseased joints and in senescent synoviocytes, along with overall m6A marks.
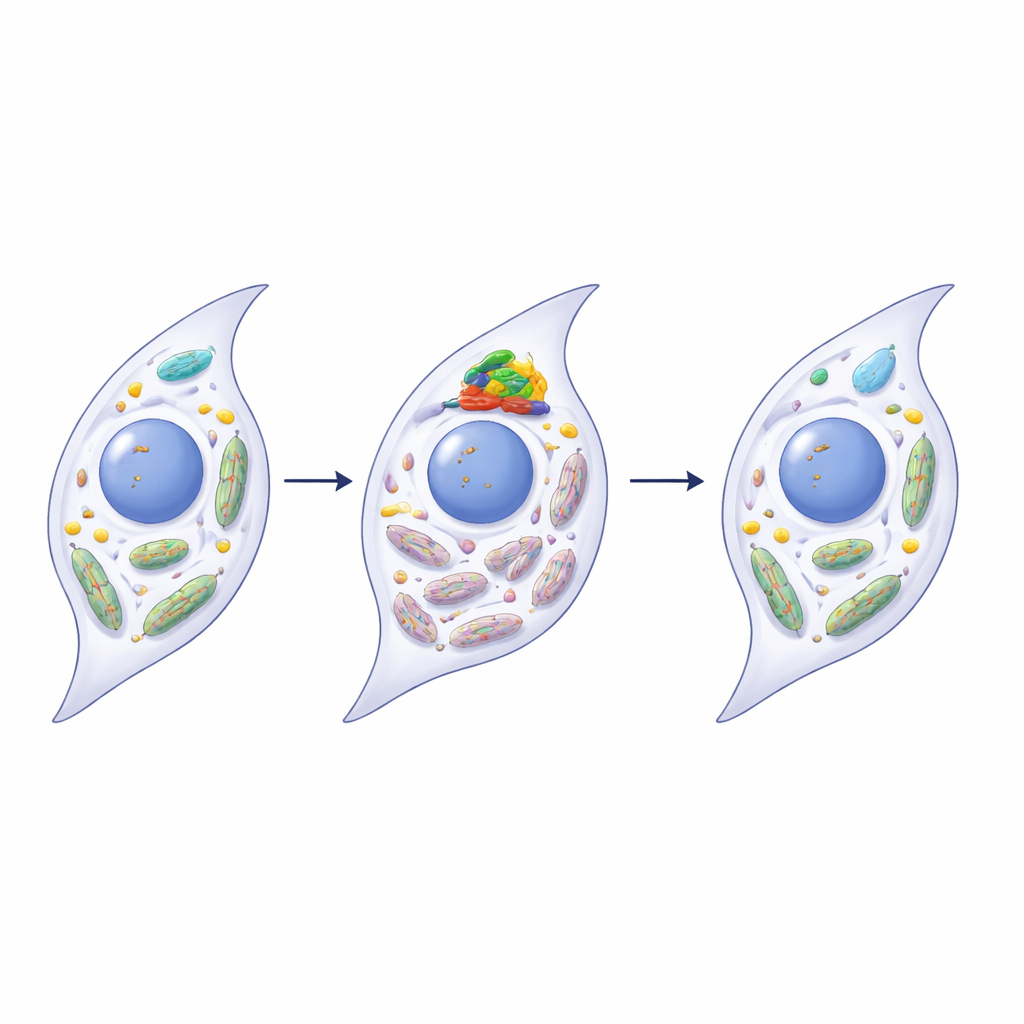
Rescuing mitochondrial health through PINK1
The researchers discovered that METTL3 directly influences a mitochondrial quality-control protein called PINK1, a central player in mitophagy. In diseased joints and in aging synoviocytes, the RNA for PINK1 carried more m6A marks and became less stable, reducing PINK1 protein levels. When METTL3 was silenced in human synoviocytes, PINK1 RNA lost some of these marks, survived longer, and produced more PINK1 protein. This, in turn, restored mitophagy, improved mitochondrial function, and reduced multiple hallmarks of senescence and inflammation. Overexpressing PINK1 alone had similar rejuvenating effects, while blocking PINK1 blunted the benefits of METTL3 silencing, tying these steps together in a single pathway from RNA modification to mitochondrial repair.
From cell aging to jaw joint damage
To see whether these aging synoviocytes could harm a joint in a living animal, the team injected either normal or senescent synoviocytes into healthy rat jaw joints. Joints that received senescent cells rapidly developed features of osteoarthritis: loss of bone quality beneath the cartilage, rough and thinned cartilage surfaces, more pain sensitivity, higher levels of inflammatory proteins, and clear reductions in markers of mitophagy. This experiment linked molecular changes inside synovial cells to full-blown joint degeneration and pain.
What this means for future jaw joint care
In plain terms, the study suggests that TMJ osteoarthritis is fueled not just by mechanical stress, but by prematurely aging joint-lining cells whose broken mitochondrial cleanup systems keep them stuck in a destructive mode. A molecular “writer” protein, METTL3, accelerates this process by destabilizing the RNA for PINK1, a key guardian of mitochondrial health. Turning down METTL3 or boosting PINK1 restored cleaner, more efficient mitochondria, calmed senescent behavior, and protected cartilage in experimental models. While these findings still need to be validated in human patients, they point to a new kind of therapy: instead of simply masking pain or replacing damaged tissue, we might slow jaw joint osteoarthritis by reprogramming how synovial cells age and maintain their internal power plants.
Citation: Tian, K., Du, Q., Guo, J. et al. METTL3-mediated fibroblast-like synoviocytes senescence promotes temporomandibular joint osteoarthritis progression. Commun Biol 9, 510 (2026). https://doi.org/10.1038/s42003-026-09773-x
Keywords: temporomandibular joint osteoarthritis, cellular senescence, mitophagy, METTL3, PINK1