Clear Sky Science · en
XMD8-92 and JWG-045 exhibit anti-ferroptotic activities, independently of inhibiting ERK5
Why protecting dying cancer cells can still teach us a lot
Cancer researchers are exploring a surprising form of cell death called ferroptosis, in which iron and damaged fats team up to punch holes in cell membranes. Because many hard‑to‑treat tumours are unusually vulnerable to this process, drugs that trigger ferroptosis are being tested as a new way to kill cancer cells. This study set out to examine whether blocking a cancer‑promoting protein, ERK5, influences how breast cancer cells respond to ferroptosis. In doing so, the researchers uncovered an unexpected twist: two widely used ERK5‑blocking chemicals actually shield cancer cells from ferroptotic death through a completely different route.
How a stress‑signalling protein meets an unusual death pathway
ERK5 is part of a signalling network that helps cells respond to growth signals and stressful conditions. Tumours with high ERK5 activity often grow faster, spread more easily, and resist other targeted drugs. A separate line of research has shown that a protective enzyme called GPX4, which normally clears harmful peroxides from cell membranes, is essential for preventing ferroptosis. When GPX4 is blocked by chemicals such as RSL3, fats in the cell membrane become oxidized, the membrane gradually breaks down, and the cell dies in a distinctive, iron‑dependent way. Because ERK5 can physically interact with GPX4, the authors wondered whether this stress‑signalling protein might make cancer cells more or less sensitive to ferroptosis.
Drugs that help and drugs that do nothing
The team first mined large cancer‑cell databases and found that tumours with high ERK5 levels tended to be more sensitive to GPX4‑blocking drugs, especially when the cells had adopted a more mobile, aggressive, so‑called mesenchymal state. They then moved to lab experiments using several breast cancer and cervical cancer cell lines. When they treated cells with RSL3 to trigger ferroptosis, two ERK5‑targeting compounds, XMD8‑92 and JWG‑045, unexpectedly kept many cells alive. Other inhibitors aimed at the same signalling pathway—newer ERK5 blockers JWG‑071 and BAY‑885, and a drug that acts one step upstream at MEK5—did not offer this protection. In all cases, the standard ferroptosis‑blocking molecule Ferrostatin‑1 behaved as expected, confirming that RSL3 was indeed killing cells through ferroptosis.
When blocking a target is not really the reason
To test whether this protective effect truly involved ERK5, the researchers reduced or completely removed ERK5 from several cancer cell lines using genetic tools, including short hairpin RNA and CRISPR gene editing. If ERK5 were essential for the anti‑ferroptotic action of these drugs, then deleting ERK5 should abolish the protection. Instead, XMD8‑92 and JWG‑045 continued to rescue cells from RSL3‑induced death even when ERK5 was barely detectable or entirely absent. In contrast, JWG‑071 and BAY‑885 still had no effect. The team also ruled out one known off‑target, the chromatin‑associated protein BRD4, because separate BRD4 inhibitors did not mimic the ferroptosis‑blocking behaviour. Together, these results pointed to a previously unrecognized off‑target activity unique to XMD8‑92 and JWG‑045.
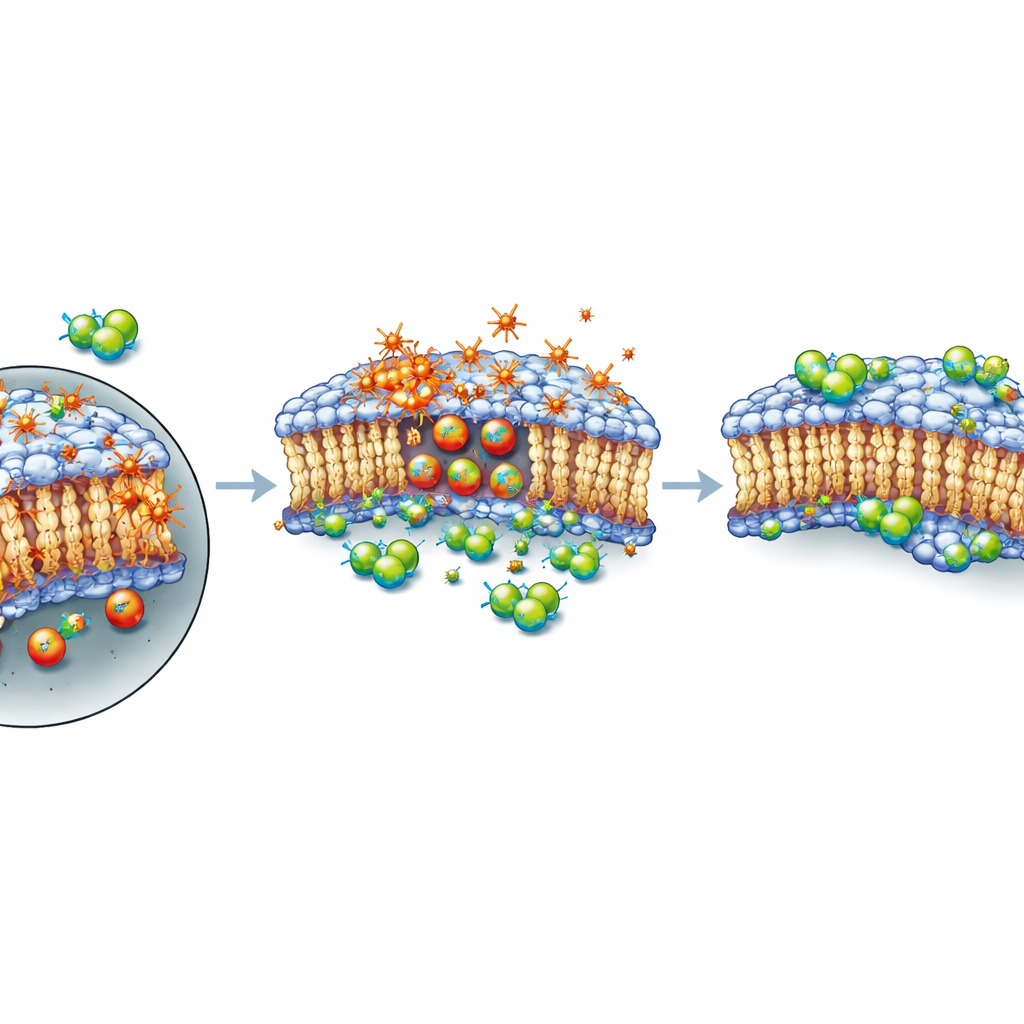
A shield for the cell’s outer skin, not a brake on damage
Next the authors asked how these compounds were intervening in the death process. By tracking gene activity over several hours after XMD8‑92 treatment, they saw only modest shifts in known ferroptosis‑related genes and no clear, coordinated change in overall ferroptosis signalling. Using a fluorescent dye that lights up oxidized fats, they showed that XMD8‑92 did not stop lipid peroxidation; if anything, oxidation increased when cells received both RSL3 and XMD8‑92. Yet cells still survived better. In carefully timed experiments, a brief exposure to RSL3 alone was enough to seal cells’ fate, even after the drug was washed away. But if XMD8‑92 was added after that short pulse, many cells recovered. This pattern suggests that the compound does not prevent the initial chemical damage; instead, it seems to help maintain or repair the plasma membrane, at least temporarily, so that cells can withstand a period of intense oxidative stress before eventually succumbing if damage continues to accumulate.
What this means for cancer research and drug design
For non‑specialists, the core message is that not all drugs behave the way their labels suggest. Two molecules widely used to study and inhibit ERK5 turn out to have an extra, powerful effect: they can delay a form of iron‑driven, membrane‑rupturing cell death without actually stopping the underlying chemical damage. This off‑target protection is important for two reasons. First, it warns researchers that results obtained with XMD8‑92 and JWG‑045 may not reliably reflect what happens when ERK5 alone is blocked. Second, it hints at the existence of drug‑accessible mechanisms that bolster membrane repair during extreme stress, which might someday be harnessed to protect healthy tissues in diseases where ferroptosis runs out of control. For now, the work underscores the need to pair newer, more selective inhibitors with genetic tools when probing ERK5 biology, and to treat correlations between gene levels and drug responses with caution rather than assuming a direct cause‑and‑effect.
Citation: Zhang, W., Kan, K., Pidd, A.B. et al. XMD8-92 and JWG-045 exhibit anti-ferroptotic activities, independently of inhibiting ERK5. Sci Rep 16, 11337 (2026). https://doi.org/10.1038/s41598-026-42079-w
Keywords: ferroptosis, ERK5 inhibitors, breast cancer, cell death pathways, drug off-target effects