Clear Sky Science · en
Network modeling of sporadic colorectal cancer reveals the importance of off-target effects of Cyclooxygenase inhibitors
Why this matters for gut health and cancer
Colorectal cancer is one of the most common cancers worldwide, and many people already take painkillers that affect the same pathways involved in this disease. This study asks a practical question with big implications: when drugs called COX inhibitors appear to slow or prevent bowel tumors, is it really because of their main intended action, or because of other, “off-target” effects inside cells? By building a detailed digital model of colon cells and their inflamed surroundings, the authors explore when and how these drugs can genuinely tip the balance from runaway growth back toward normal cell turnover.
Untangling a crowded signal landscape
Colon tumors do not arise from a single damaged gene but from a tangled web of signals between gut cells, immune cells, and inflammatory molecules. The researchers assembled a large on–off (Boolean) network that captures 87 key players inside a typical intestinal cell and its nearby immune environment. These include well-known growth drivers, brakes on cell death, and molecules released during inflammation. They then allowed this virtual network to run thousands of times, tracking how often the “proliferation” node (cell division) and the “apoptosis” node (programmed cell death) ended up switched on. This approach let them test how different conditions—such as chronic inflammation or common cancer-linked gene changes—shift the overall behavior of the system.
When inflammation locks cells in growth mode
The first surprise came when the team turned on the inflammatory environment in their model. Signals such as IL6 and CCL2, made by immune and epithelial cells, formed two reinforcing feedback loops that strongly boosted growth pathways and shut down cell death. If these signals were allowed to act at full strength, the model colon cells became almost endlessly proliferative and nearly impossible to kill—far from what is seen in healthy tissue. By dialing down the probability that IL6 and CCL2 would turn on, the authors found a sweet spot where cells kept a low, normal growth rate but could still respond to inflammatory triggers. Persistent activation of certain immune cells produced a chronic state that lowered cell death and slightly raised growth, mimicking how long-term inflammation can make the gut lining more vulnerable to cancer.
How common gene changes push cells over the edge
Next, the researchers introduced genetic alterations often seen in sporadic colorectal cancer, such as loss of the APC “gatekeeper” gene, activating changes in RAS, and loss of the guardian gene p53. In the model, each of these changes, in its own way, fed into the same two positive feedback loops. The result was a clear shift toward survival and proliferation: cell death signals dropped, while growth signals surged. By contrast, simulated loss of a chromosome region containing SMAD and DCC—alterations tied to poor prognosis in real patients—barely changed proliferation or apoptosis in the model, suggesting that pathways controlled by these genes were underrepresented and may need to be modeled in more detail to match clinical reality.
Off-target actions make or break drug benefit
The heart of the study was to see how blocking different drug targets would affect this network, especially targets hit by common COX inhibitors such as celecoxib and sulindac. When the only problem in the simulated cell was loss of APC, directly blocking COX2 or a related enzyme (PDE5) fully restored a healthy balance: growth dropped and cell death rose above normal, breaking the feedback loops early. But as additional mutations in RAS and p53 were layered in—representing intermediate and late tumor stages—simply blocking COX2 did little. In those later settings, inhibiting the growth pathway centered on AKT, or to a lesser degree NF-κB, was far more effective at slowing proliferation, though it still could not fully revive cell death. The model also suggested that celecoxib, which can hit AKT as well as COX2, should out-perform sulindac in restraining tumor cell growth when multiple mutations are present.
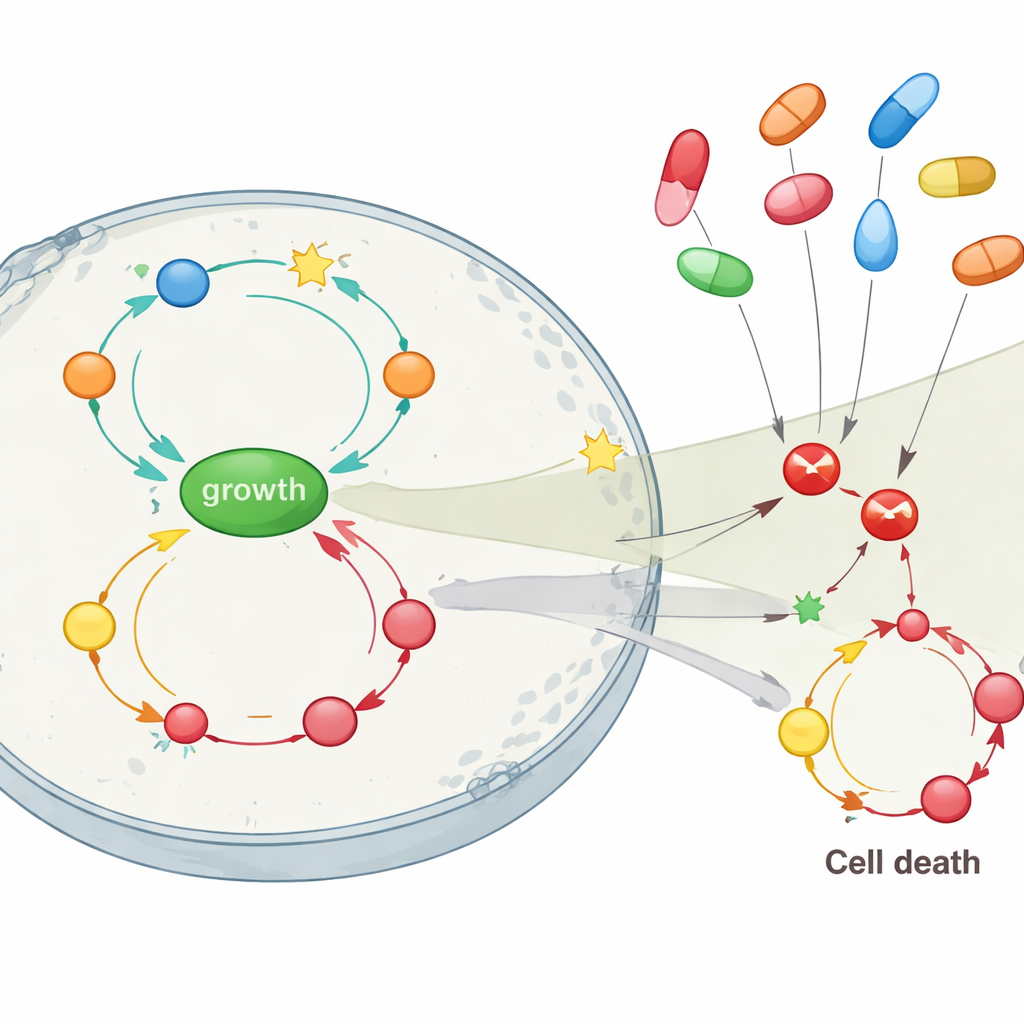
What this means for prevention and treatment
For a layperson, the key message is that COX inhibitors do not work as simple, single-target drugs against colon cancer. Their helpful effects, especially in early disease, may rely heavily on additional actions against other growth pathways. The study’s simulations suggest that these drugs are most powerful when used before tumors accumulate many mutations—particularly when the crucial APC gatekeeper gene is lost but other changes have not yet locked growth circuits fully on. In more advanced cancers, blocking COX2 alone is unlikely to be enough, and attention may need to shift toward pathways like AKT. By mapping these complex feedback loops in silico, the work supports a more tailored approach: using genetic information and inflammation status to decide who might benefit from preventive COX inhibitor use, and when combination strategies targeting multiple nodes in the network may be required.
Citation: Gebhart, A.R., Berns, M.M.M., Snoeys, J. et al. Network modeling of sporadic colorectal cancer reveals the importance of off-target effects of Cyclooxygenase inhibitors. npj Syst Biol Appl 12, 50 (2026). https://doi.org/10.1038/s41540-025-00622-x
Keywords: colorectal cancer, inflammation, COX inhibitors, signal networks, AKT pathway