Clear Sky Science · en
Recruitment of BRD4 to the ASXL1 genomic targets depends on the extra-terminal domain of BRD4
Why this study matters
Cancer often hijacks the cell’s gene switches, turning growth programs on when they should be off. Two such key gene regulators, ASXL1 and BRD4, are frequently altered or overactive in blood cancers and brain tumors. This study uncovers, in molecular detail, how these two proteins physically connect on DNA, how cancer-promoting forms of ASXL1 strengthen that connection, and why this partnership may be an attractive target for new cancer therapies. 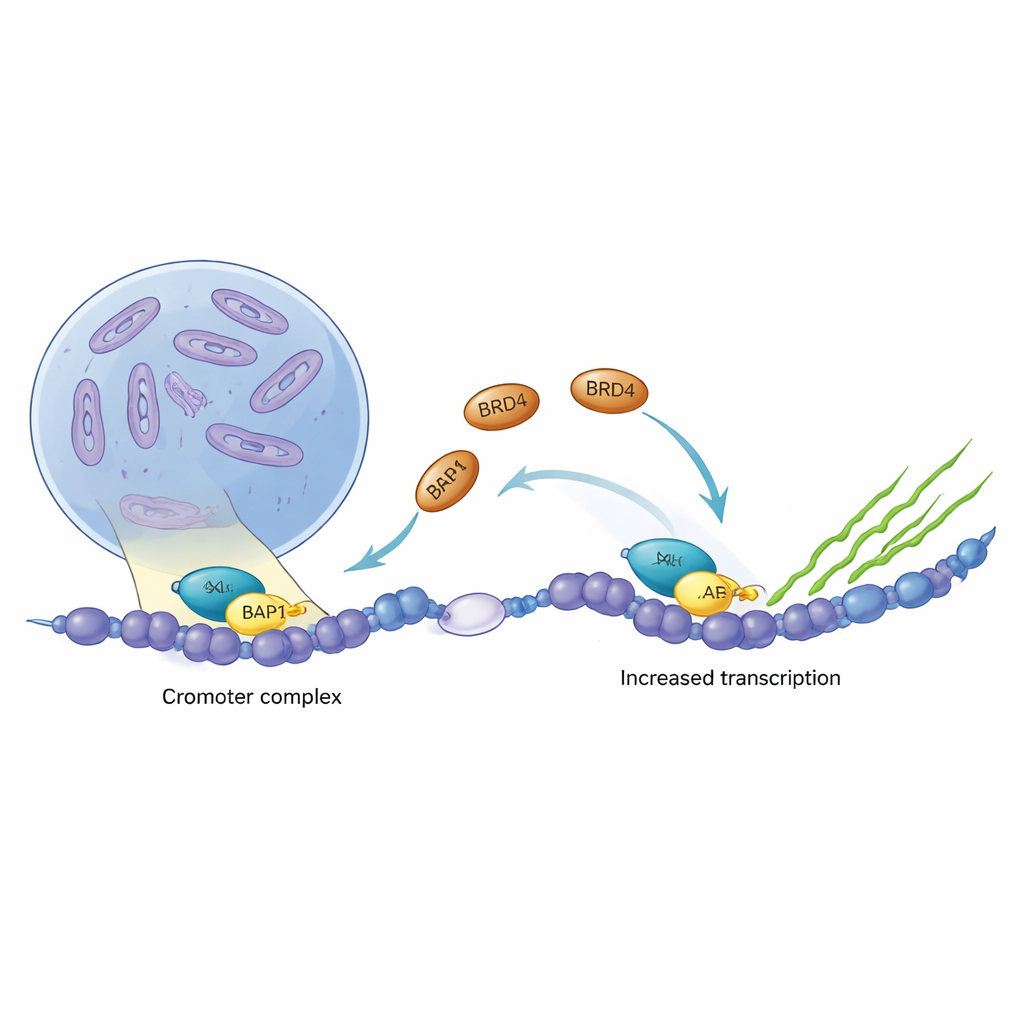
A partnership at the heart of gene control
ASXL1 is part of a protein crew that fine-tunes how tightly DNA is packed, helping decide which genes are silent and which are active. BRD4, in contrast, acts as a powerful ignition switch for transcription, the process that reads genes into RNA. By analyzing cancer data from several tumor types, the authors found that ASXL1 and BRD4 tend to rise and fall together, and that BRD4 frequently occupies the DNA region that drives ASXL1’s own production. This tight correlation hinted that the two proteins might collaborate directly to boost specific gene programs in cancer cells.
Zooming in on the molecular handshake
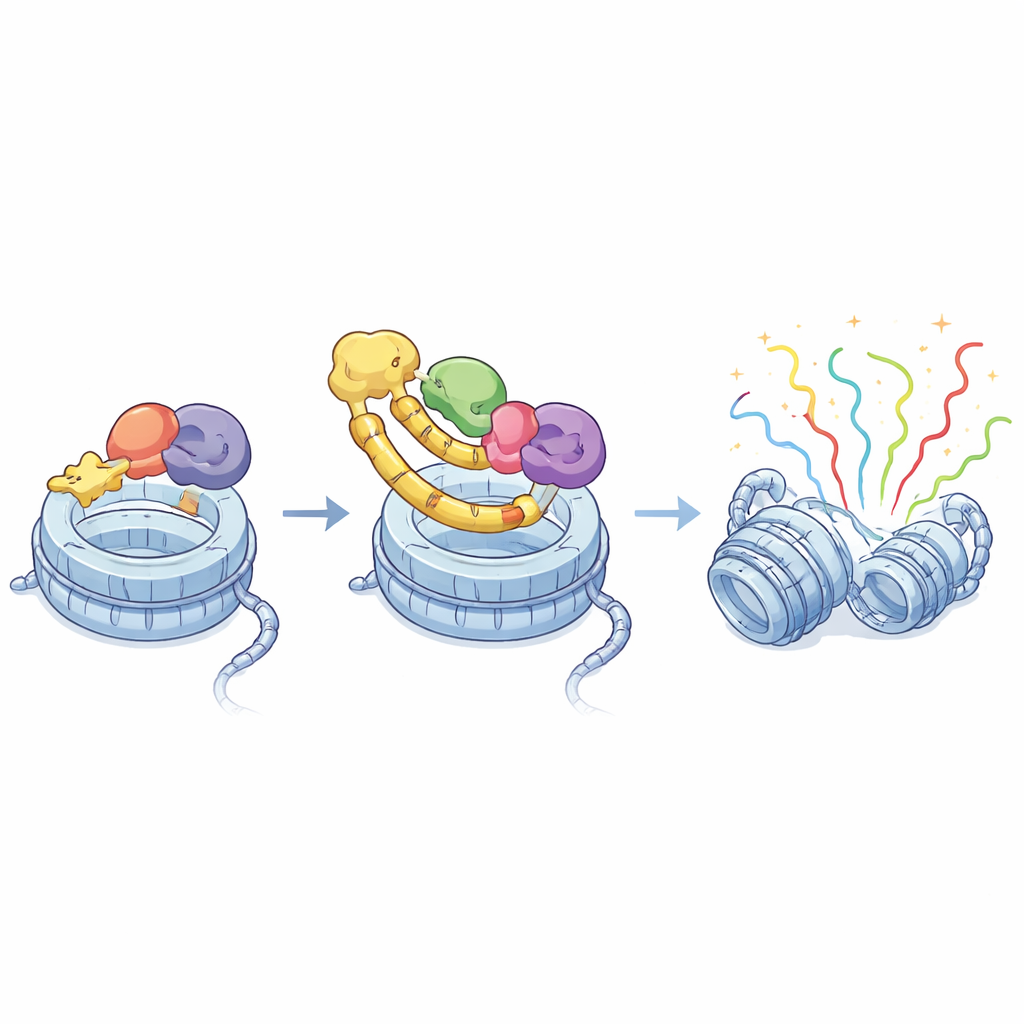
To understand how ASXL1 and BRD4 connect, the team used high-resolution structural tools and biochemical tests. They showed that a small stretch within ASXL1 fits snugly into a groove in BRD4’s so‑called extra‑terminal (ET) region, forming a tight complex. Nuclear magnetic resonance experiments revealed that ASXL1 contributes a short, extended strand that pairs with part of BRD4, while hydrophobic and charged side chains interlock like teeth in a zipper. Mutating just one or two of these key ASXL1 or BRD4 residues was enough to largely abolish binding, proving that the interaction is highly specific and structurally well defined. Intriguingly, ASXL1’s close relatives, ASXL2 and ASXL3, use similar short motifs to bind the same ET groove, suggesting a conserved recognition code.
How cancer-linked truncations reshape the complex
Many patients with myeloid blood cancers carry truncated versions of ASXL1 that end roughly halfway through the protein. The authors focused on two common forms that stop at positions 591 and 645. Both shorten the protein but preserve the BRD4‑binding region. When these variants were introduced into human cells, they pulled down BRD4 just as effectively as the normal protein, unless a single critical contact residue was altered. Genome‑wide binding maps showed that BRD4 is strongly enriched at promoters where ASXL1 is present, and this enrichment drops when the ASXL1–BRD4 contact is disrupted. Surprisingly, the slightly longer truncation (ending at 645) was better at bringing BRD4 to certain gene promoters than the shorter one, indicating that losing some parts of ASXL1 can paradoxically create a stronger, cancer-fueling bridge to BRD4.
A three-way hub for activating genes
From molecular insight to treatment ideas
By combining structural biology, genome mapping, and cancer genomics, the study shows that BRD4’s ET region physically latches onto a short motif in ASXL1 and that this “molecular handshake” is essential for guiding BRD4 to ASXL1‑controlled genes. Cancer-associated ASXL1 truncations not only preserve this handshake but, in some cases, enhance BRD4 recruitment and add the ability to tether MLL3/4, amplifying gene activation. Because these interactions rely on a well‑defined surface on BRD4 and a short stretch of ASXL1, they provide a clear blueprint for designing small molecules that could selectively disrupt the ASXL1–BRD4 link, potentially damping cancer-driving gene programs while sparing other BRD4 functions.
Citation: Selvam, K., Lu, S., Messmer, C. et al. Recruitment of BRD4 to the ASXL1 genomic targets depends on the extra-terminal domain of BRD4. Nat Commun 17, 2852 (2026). https://doi.org/10.1038/s41467-026-69565-z
Keywords: epigenetic regulation, BRD4, ASXL1, chromatin complexes, cancer transcription